Я хочу прекратить деятельность ИП | ФНС России
Содержание страницыФормируем пакет документов
Вам потребуются следующие документы:
заявление о государственной регистрации прекращения физическим лицом деятельности в качестве индивидуального предпринимателя в связи с принятием им решения о прекращении данной деятельности (форма № Р26001)
Подпись на заявлении должна быть засвидетельствована в нотариальном порядке, за исключением случая, когда заявитель представляет документы лично и одновременно представляет паспорт.
квитанция об уплате госпошлины в размере 160 руб.
Перейти Сформировать квитанцию на уплату госпошлины с помощью сервиса: «Уплата госпошлины»документ, подтверждающий представление сведений в территориальный орган Пенсионного фонда.

Документ, подтверждающий представление сведений в территориальный орган Пенсионного фонда, не обязателен. Если заявитель не представит этот документ, нужную информацию территориальный орган Пенсионного фонда направит налоговому органу в электронном виде в рамках межведомственного обмена.
Перечень сведений, представляемых в территориальный орган Пенсионного фонда, определен подп. 1–8 п. 2 ст. 6 и п. 2 ст. 11 Федерального закона от 01.04.1996 № 27-ФЗ «Об индивидуальном (персонифицированном) учете в системе обязательного пенсионного страхования», а также ч. 4 ст. 9 Федерального закона от 30.04.2008 № 56-ФЗ «О дополнительных страховых взносах на накопительную часть трудовой пенсии и государственной поддержке формирования пенсионных накоплений».
Представляем документы
Документы могут быть переданы в налоговую инспекцию любым удобным для вас способом:
- непосредственно в инспекцию — лично или через представителя по доверенности.

- в многофункциональный центр — лично или через представителя по доверенности. Информацию об оказании данной услуги в Вашем МФЦ необходимо уточнить на сайте МФЦ.
- по почте с объявленной ценностью и описью вложения;
В пределах территории Москвы документы можно направить и получить также через DHL Express и Pony Express.
в электронном виде.
Подать документы Подать документы с помощью сервиса:
заявки на государственную регистрацию индивидуальных предпринимателей и юридических лиц»
Инспекция примет документы и выдаст (направит) расписку в их получении.
Получаем документы
На 6-й рабочий день после подачи документов заявитель лично или через представителя по нотариально удостоверенной доверенности может получить:
- лист записи ЕГРИП
В случае отказа в государственной регистрации вы получите документ, в котором изложена причина отказа.
Перечень оснований для отказа в государственной регистрации определен п. 1 ст. 23 Федерального закона от 08.08.2001 № 129-ФЗ «О государственной регистрации юридических лиц и индивидуальных предпринимателей».
Документ могут направить в ваш адрес и по почте. В пределах территории Москвы документ можно получить также через DHL Express и Pony Express.
ИП закрывается: что можно и нужно делать
Индивидуальный предприниматель, решивший прекратить деятельность, может закрыть ИП самостоятельно. Требуемый алгоритм действий приведен в отдельном материале. Но бизнесмен также вправе обратиться для выполнения услуги по закрытию ИП в компанию «Такском». Специалисты проведут процедуру ликвидации компетентно и быстро. А сейчас отвечаем на часто задаваемые вопросы, возникающие при закрытии ИП.
Специалисты проведут процедуру ликвидации компетентно и быстро. А сейчас отвечаем на часто задаваемые вопросы, возникающие при закрытии ИП.
Можно ли закрыть ИП с долгами перед государством?
Индивидуальный предприниматель может закрыть ИП с задолженностью Пенсионному фонду. В этом случае он будет выплачивать этому госоргану долг после прекращения предпринимательской деятельности.
А вот непогашенные обязательства перед ФНС не позволят ликвидировать ИП. Сначала нужно выплатить задолженность, причем, вместе со штрафами и пенями, а потом закрывать ИП. Перед тем, как начать процедуру ликвидации, необходимо подготовить отчеты за весь период деятельности предпринимателя даже в том случае, если ее как таковой не было.
Как оплачивать долги перед госорганами после закрытия ИП?
Пенсионный фонд должен быть автоматически оповещен о снятии предпринимателя с учета в налоговой инспекции. Но это по какой-либо технической причине может не произойти. Поэтому ИП лучше подстраховаться и самому оповестить ПФР сразу после ликвидационной процедуры и затем оплатить требуемые страховые взносы, как можно быстрее, желательно не позднее 15 календарных дней с момента получения документов о закрытии бизнеса. Сотрудник ПФР предоставит все необходимые для этого квитанции.
Можно ли после закрытия ИП его открыть снова?
Можно, но имеет значение, как был закрыт ИП. В том случае, если ИП был признан решением арбитражного суда банкротом, он не вправе заниматься предпринимательской деятельностью в течение срока, установленного статьей 216 Федерального закона от 26.10.2002 № 127-ФЗ в редакции от 31.07.2020. Срок зависит от конкретной ситуации и составляет от одного года до пяти лет.
Кроме того, 1 сентября 2020 года вступила в силу еще одна интересная норма, зафиксированная в Федеральном законе от 08.08.2001 № 129-ФЗ. Она позволяет ФНС самостоятельно закрыть ИП, исключив его из ЕГРИП, если он:
— в течение 15 месяцев не подавал налоговые отчёты, расчёты и не продлевал патент;
— имеет недоимку и задолженность, в которую могут входить неуплаченные штрафы и пени, по налогам и сборам.
После исключения из ЕГРИП по решению регистрирующего органа согласно новой редакции пункта 4 статьи 22. 1 Федерального закона № 129-ФЗ физическое лицо не вправе вновь зарегистрировать ИП в течение трех лет.
1 Федерального закона № 129-ФЗ физическое лицо не вправе вновь зарегистрировать ИП в течение трех лет.
Налогоплательщик, прекративший деятельность в качестве ИП добровольно и не имеющий долгов перед государством, может открыть ИП в любой момент.
Как правильно уволить сотрудников при ликвидации ИП?
После принятия решения о закрытии собственного бизнеса предприниматель – работодатель должен уведомить сотрудников о предстоящем увольнении под роспись не менее чем за два месяца до начала ликвидации.
Кроме того, он обязан не позже чем за две недели до старта процедуры и не позднее, чем за три месяца при массовом увольнении персонала в письменной форме уведомить об этом службу занятости.
Далее до исключения ИП из ЕГРИП издается на каждого из наемных работников приказ об увольнении на основании пункта 1 статьи 81 ТК РФ в связи прекращением деятельности. Компенсации и выходные пособия выплачиваются только, если это условие отражено в трудовом договоре. В день увольнения выдается трудовая книжка и справка о заработке для расчета пособий за текущий и два предыдущих года, а также форма 2-НДФЛ за текущий год.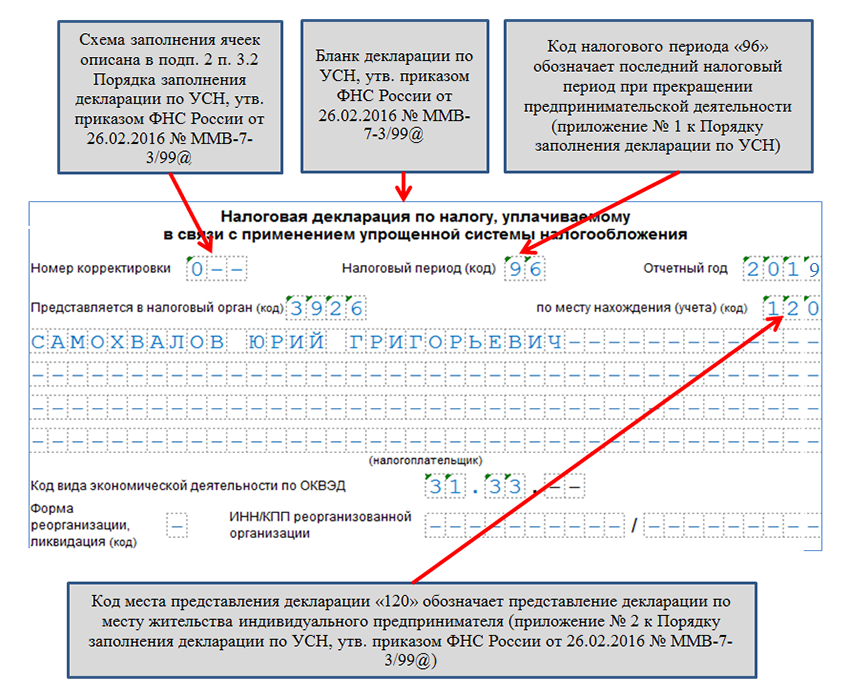
По уволенным сотрудникам предприниматель должен представить следующую отчетность:
— Расчет по страховым взносам и 6-НДФЛ – в налоговую инспекцию;
— СЗВ-М, СЗВ-СТАЖ и СЗВ-ТД – в Пенсионный фонд;
— 4-ФСС – в Фонд социального страхования.
Взносы, исчисленные в расчете по страховым взносам и отчете 4-ФСС ИП должен уплатить в течение 15 календарных дней с момента подачи расчета.
Нужно ли снимать кассу с учёта при закрытии ИП?
При прекращении деятельности, в которой ИП использовал контрольно-кассовую технику, логично возникает вопрос о снятии ее с учета. И если ИП намеревается это сделать сам, ему следует подать заявление в налоговую инспекцию и приложить отчет о закрытии фискального накопителя. Заявление он может направить как в бумажном, так и электронном виде. И в том и другом случае касса будет снята с учета в течение 10 рабочих дней с даты подачи заявления.
Однако поправки, внесенные в Федеральный закон № 54-ФЗ, значительно упростили процедуру «закрытия» кассы. Согласно им у налоговых органов появилось право снимать ККТ с учета, если в ЕГРИП, а также и в ЕГРЮЛ внесена запись о ликвидации ИП или компании. Таким образом, получается, что как только в ЕГРИП попадет информация о том, что предпринимательская деятельность прекращена, ИФНС самостоятельно снимет кассу с учета. В этой ситуации единственное, что нужно ИП сделать, — убедиться, что на момент выключения ККТ все фискальные документы переданы ИФНС.
Согласно им у налоговых органов появилось право снимать ККТ с учета, если в ЕГРИП, а также и в ЕГРЮЛ внесена запись о ликвидации ИП или компании. Таким образом, получается, что как только в ЕГРИП попадет информация о том, что предпринимательская деятельность прекращена, ИФНС самостоятельно снимет кассу с учета. В этой ситуации единственное, что нужно ИП сделать, — убедиться, что на момент выключения ККТ все фискальные документы переданы ИФНС.
Каким образом закрыть расчётный счёт при ликвидации ИП?
Счет в банке закрывается после ликвидации ИП. Перечень документов, требуемый для этого, есть в договоре на обслуживание. Обычно — это заявление на закрытие, паспорт, выписка из ЕГРИП. После того, как ИП собрал все необходимые документы, он должен получить выписку о состоянии счета, погасить все долги, штрафы, пени и вывести оставшиеся средства. Он может перевести их на карту или получить в кассе. И уже после этого он должен подать заявление на закрытие по утвержденной в конкретном банке форме.
Кредитная организация после получения заявления проверяет физическое лицо на предмет долговых обязательств. И если задолженности не выявляет, направляет бывшему клиенту уведомление о расторжении договора с банком. Далее выдается справка о закрытии расчетного счета.
В течение какого времени надо хранить документы после закрытия ИП?
Минимальные сроки хранения отдельных категорий документов ИП после его закрытия:
— первичные документы и бухгалтерские бумаги, необходимые для исчисления и оплаты налогов, — четыре года;
— книга учета доходов и расходов (КУДИР) и другие документы, подтверждающие доходы и расходы, — четыре года;
— иная бухгалтерская отчётность — пять лет;
— отчётность по административно-хозяйственным вопросам — пять лет;
— документация, подтверждающая исчисление и оплату страховых взносов в ФСС, ФФОМС и ПФР, — шесть лет.
Закрыть ИП – без нервов и суеты
✔ от 1 до 5 рабочих дней;
✔ без посещения налоговой и офиса «Такскома»;
✔ конфиденциально, абсолютно законно
Заказать услугу
Отправить
Запинить
Твитнуть
Поделиться
Поделиться
Способы подачи заявления на ликвидацию ИП
Онлайн
В сервисе «Документовед» можно подготовить документы для электронной подачи.
Для этого нужно заполнить анкету на Ликвидацию ИП.
После отправляем заявку на формирование контейнера. Наши специалисты формируют для вас контейнер для ликвидации ИП.
Заявление отправляется в налоговую и в течение следующего дня должна прийти расписка.
Получение документов
Ликвидацию ИП налоговая зарегистрирует в течение 5 рабочих дней.
Лист записи о прекращении деятельности индивидуального предпринимателя ФНС отправит на электронную почту, указанную в форме Р26001. Он будет подписан ЭЦП налогового органа и в соответствии с Федеральным законом № 63-ФЗ «Об электронной подписи» равнозначен документу с проставленной синей печатью.
При желании Вы можете получить лист записи в ЕГРИП на бумажном носителе способом доставки, указанном в заявлении (лично либо почтой).
Преимущества и недостатки онлайн подачи
Основные плюсы электронной подачи через сервис «Документовед»:
- Без личного посещения налоговой инспекции.
- Можно подать документы, не выходя из дома.
- Ваше местоположение не важно — заявление поступит в нужную налоговую инспекцию.
- Без оплаты государственной пошлины и услуг нотариуса.
Единственный недостаток данного способа — для онлайн подачи потребуется квалифицированная электронная подпись.
Лично
Пока самым распространённым способом является личная подача заявления в налоговую инспекцию. Прекращение деятельности ИП осуществляется в регистрирующей ФНС по месту его жительства, то есть по месту регистрации, указанному в паспорте.
Уточните режим работы инспекции, а также нужна ли предварительная запись.
Важно! При личной подаче в ФНС не забудьте паспорт.
Если электронной записи нет, то по приходу в налоговую найдите окошко для приема заявления на ликвидацию, и займите очередь.![]()
Проверьте пакет документов, необходимый для подачи:
- заполненная форма Р26001;
- квитанция на уплату пошлины;
- справка, подтверждающая представление сведений в ПФР (не обязательно).
Инспектор налоговой обязан принять все документы, попросить поставить в его присутствии подпись на форме Р26001 и выдать расписку.
На расписке будет указана дата готовности — через 5 рабочих дней со дня, следующего за днем подачи.
Если в Р26001 было отмечено личное получение, то с паспортом и распиской вернитесь в налоговую и получите лист записи в ЕГРИП. Если отметили получение по почте — документ, подтверждающий закрытие ИП, будет отправлен на домашний адрес.
Преимущества и недостатки личной подачи в ФНС
Из преимуществ можно отметить отсутствие необходимости заверения формы, оформления доверенности. Также этот способ надежнее отправки заявления по почте.
Но есть ряд существенных недостатков:
- существенная потеря времени на посещение налоговой;
- необходимо будет оплачивать государственную пошлину;
- часто регистрирующая налоговая находится на большом расстоянии от места жительства.

А если гражданин зарегистрирован в одном регионе, а проживает совсем в другом месте, и у него нет возможности доехать до нужной налоговой. Тогда единственным вариантом будут удаленные способы подачи документов.
МФЦ
Прежде чем подавать документы на закрытие ИП в МФЦ, нужно выяснить, в каком из них это можно сделать — такая услуга есть не во всех центрах государственных услуг.
При посещении МФЦ не забудьте взять с собой паспорт!
В присутствии специалиста МФЦ поставьте подпись на заявлении Р26001 и получите расписку.
Срок регистрации ликвидации при таком способе подачи немного увеличивается (7 рабочих дней), так как нужно пару дней для передачи документов из МФЦ в налоговую и обратно.
При получении результата ликвидации сотрудник МФЦ проверит Ваш паспорт и выдаст бумажный документ, подтверждающий содержание электронного листа записи.
Преимущества и недостатки личной подачи в МФЦ
Из преимуществ можно отметить отсутствие необходимости оформления доверенности. Также при подаче в МФЦ не нужно оплачивать государственную пошлину, т.к. документы в ФНС будут передаваться в электронном виде.
Главный из недостатков — далеко не все центры государственных услуг могут предоставить данную услугу. Также предоставить заявление Вы можете не в любой МФЦ, а только по месту своего проживания.
По доверенности
При таком способе подачи нужно будет посетить нотариуса для заверения подписи в форме Р26001 и оформления доверенности.
Порядок подачи через представителя не отличается от личной подачи. Все действия по подаче и получению документов ложатся на плечи представителя.
Преимущества и недостатки подачи через представителя
Из преимуществ можно отметить отсутствие необходимости посещения налоговой лично Вами. В налоговую отправится Ваш представитель.
Основной недостаток — посещение нотариуса:
- Это может отнять много времени, так как чаще всего у нотариусов очередь, даже если Вы предварительно записывались.

- Стоимость нотариальных услуг. Заверение формы —1500 -2000 ₽, оформление доверенности —1500 -2000 ₽.
По почте
Для отправки по почте заявление Р26001 предварительно нужно будет заверить у нотариуса.
После отправляемся в любое почтовое отделение.
1. Заполняем конверт. На конверте внимательно вписываем:
- КОМУ: Наименование регистрирующего налогового органа и полный почтовый адрес;
- ОТ КОГО: Вписываем свой адрес, куда налоговый орган отправит расписку, а потом при положительном решении — документы, подтверждающие закрытие ИП.
2. Вкладываем в конверт необходимые бумаги.
3. Оформляем опись вложения в двух экземплярах.
4. Оформляем уведомление о вручении (обязательно регистрируемое).
5. Конверт с вложенными документами, опись вложения в двух экземплярах и заполненный бланк регистрируемого уведомления передаём оператору почтовой связи.
6. Предупреждаем оператора, что необходимо оформить отправление с объявленной ценность, описью вложения и уведомлением.
7. Оператор выдаст один экземпляр описи вложения и чек с трек-номером, для возможности отслеживания прохождения отправления на сайте ФГУП Почта России.
Получение документов
После того, как налоговая получит Ваше отправление, обработает его, отправит на указанный Вами адрес расписку.
Момент поступления заявления на закрытие ИП в налоговую Вы можете отслеживать по трек-номеру в чеке, который выдал оператор на почте.
В случае успешной регистрации, лист записи в ЕГРИП отправляют по почте, если Вы указали именно такой способ получения документов.
Преимущества и недостатки
Из преимуществ можно отметить отсутствие необходимости посещения налоговой. Документы можно отправить в любом почтовом отделении, независимо от места Вашей постоянной регистрации.
Недостатков такого способа значительно больше.
Основной недостаток — расходы на заверение подписи в заявлении Р26001 (1500-2000 ₽).
Также необходимо будет оплатить почтовые услуги.
И самое важное, отправка почтой России — достаточно долгий и ненадежный способ закрытия ИП. Документы могут идти в налоговую около недели, а могут затеряться и до налоговой не дойти.
Закрытие ИП
Чтобы предпринимателю закрыть своё ИП, нужно пройти ряд этапов.
Важное правило: вы можете подавать документы на закрытие ИП, даже имея долги перед ИФНС, ПФР или другими лицами или несданную отчетность. Дело в том, что закрытие ИП не прекращает возникшие у человека долги, их все придется закрывать даже после прекращения деятельности. Поэтому, они не мешают подаче документов на прекращение деятельности. Регистрация прекращения деятельности ИП в налоговой длится 5 рабочих дней.
Однако, прежде чем закрываться, мы рекомендуем выяснить состояние долгов в ПФР и ИФНС — это сделает закрытие более предсказуемым.
После закрытия ИП нужно сдать финальную налоговую декларацию (УСН или 3-НДФЛ).
Есть нюанс — доверенное лицо (поверенный) может представлять интересы индивидуального предпринимателя — физического лица, только на основании нотариальной доверенности. Из этого следует, что либо ИП будет самостоятельно посещать все госорганы, либо достаточно один раз посетить нотариуса, заверив положенные документы и выписав нам нотариальную доверенность. Тогда мы всё сделаем за вас.
Сколько времени это займет (план наших действий)
| Наши действия | Сроки |
|---|---|
| Подготовка документов на закрытие ИП | 1 день |
| Подача документов в МИФНС 46 | 1 день |
| Рассмотрение документов в МИФНС 46 | 5 дней |
| Получение результата | 1 день |
| Сдача окончательной налоговой декларации в ИФНС | 1 день |
| Всего «под ключ» в стандартном варианте | 9 рабочих дней |
Стоимость услуги по закрытию ИП
| Наши услуги | Цена |
|---|---|
| Подготовка документов на закрытие ИП Подача-получение в МИФНС 46 нашим специалистом по доверенности |
6000 |
| Подготовка и сдача финальной налоговой декларации в ИФНС (нулевая / ненулевая) | 4000 / по договоренности |
| Решение вопросов с долгами ПФР (при необходимости) | от 10000 р |
| Накладные расходы | |
| Государственная пошлина за прекращение деятельности индивидуального предпринимателя | 160 |
Нотариальные расходы (если подаете по доверенности нашим курьером):
|
примерно: 1400 1000 1500 240 ——— 4140 |
Вы можете заказать услугу «под ключ» или же выбрать отдельные этапы, которые актуальны именно вам.
После закрытия ИП вы получите на руки полный пакет документов:
- Лист записи о прекращении деятельности, с отметкой МИФНС 46.
- Декларация УСН или НДФЛ с подтверждением электронной сдачи.
Часто задаваемые вопросы
Можно ли перерегистрировать ИП в ООО?
Нет, такой процедуры законодательством не предусмотрено. Если гражданин хочет осуществлять предпринимательскую деятельность от имени ООО, а не от имени ИП, ему следует закрыть ИП и открыть ООО.
Нужно ли платить налоги и взносы после закрытия ИП?
Прекращение регистрации индивидуального предпринимателя не влечет автоматического прекращения его обязательств по уплате налогов. Эти обязательства прекращаются только в случаях:
— уплаты налога или сбора
— в связи со смертью физического лица (ст. 44 НК РФ)
Поэтому, если какие-то налоги за время работы в качестве ИП у вас остались неуплаченными, их нужно доплатить.
Можно ли оштрафовать ИП после прекращения деятельности?
Да, так как после закрытия ИП все права и обязанности не прекращаются, а остаются у физического лица.
Сколько лет нужно хранить документы по закрытому ИП?
Документы следует хранить в течение полных четырех лет.
Типичные ошибки и их последствия
Забыли сдать окончательную декларацию
Прекращение регистрации в качестве индивидуального предпринимателя не снимает с физического лица обязанность сдать декларации. За несдачу декларации положен штраф.
Есть вопросы?
Задайте их нам через форму обратной связи прямо сейчас.
-
Введите Ваше имя*
Введите имя.
-
E-mail*
Не верный адрес e-mail.
-
Телефон
-
Текст вашего сообщения
Неверный ввод
-
Проверка на спам
Неверный ввод -
Отправить Очистить
Ошибка: 404 — Страница не найдена
Настоящим в соответствии с Федеральным законом №152‑ФЗ
«О персональных данных» от 27. 07.2006 года свободно, своей волей и в своём
интересе выражаю своё безусловное согласие на обработку моих персональных данных
НАЗВАНИЕ КОМПАНИИ,
зарегистрированным в соответствии с законодательством РФ по адресу:
АДРЕС КОМПАНИИ
(далее по тексту — Оператор).
07.2006 года свободно, своей волей и в своём
интересе выражаю своё безусловное согласие на обработку моих персональных данных
НАЗВАНИЕ КОМПАНИИ,
зарегистрированным в соответствии с законодательством РФ по адресу:
АДРЕС КОМПАНИИ
(далее по тексту — Оператор).
Персональные данные — любая информация, относящаяся к определённому или определяемому на основании такой информации физическому лицу.
Настоящее Согласие выдано мною на обработку следующих персональных данных:
- Имя;
- Телефон;
- E-mail;
-
Комментарий.

Согласие дано Оператору для совершения следующих действий с моими персональными данными с использованием средств автоматизации и/или без использования таких средств: сбор, систематизация, накопление, хранение, уточнение (обновление, изменение), использование, обезличивание, передача третьим лицам для указанных ниже целей, а также осуществление любых иных действий, предусмотренных действующим законодательством РФ, как неавтоматизированными, так и автоматизированными способами.
Данное согласие даётся Оператору и третьему лицу(‑ам) ТРЕТЬИ ЛИЦА для обработки моих персональных данных в следующих целях:
- предоставление мне услуг/работ;
- направление в мой адрес уведомлений, касающихся предоставляемых услуг/работ;
- подготовка и направление ответов/коммерческих предложений на мои запросы;
-
направление в мой адрес информации, в том числе рекламной,
о мероприятиях/товарах/услугах/работах Оператора.

Настоящее согласие действует до момента его отзыва путём направления соответствующего уведомления на электронный адрес ЕМЕЙЛ. В случае отзыва мною согласия на обработку персональных данных Оператор вправе продолжить обработку персональных данных без моего согласия при наличии оснований, указанных в пунктах 2‑11 части 1 статьи 6, части 2 статьи 10 и части 2 статьи 11 Федерального закона №152‑ФЗ «О персональных данных» от 26.06.2006 г.
Как открыть закрытое ИП — порядок действий
Когда можно открыть закрытое ИП?
Прекращение предпринимательской деятельности происходит по двум сценариям. Основанием для первого процесса является добровольное желание ИП закрыть свой бизнес в связи с низким доходом или желанием заниматься другим делом. Вторая причина – принудительная остановка деятельности по решению суда. В зависимости от того, по какому сценарию происходит закрытие ИП, будет зависеть, какие вопросы предстоит решать предпринимателю в будущем при открытии бизнеса снова.
Вторая причина – принудительная остановка деятельности по решению суда. В зависимости от того, по какому сценарию происходит закрытие ИП, будет зависеть, какие вопросы предстоит решать предпринимателю в будущем при открытии бизнеса снова.
На практике чаще встречаются случаи добровольного прекращения деятельности бизнесмена.
Порядок закрытия
Если у гражданина не было работников по трудовому договору, то прекращение работы ИП имеет следующий порядок:
- составление заявления с просьбой снятия с учета в ЕГРИП;
- оплата госпошлины;
- подача пакета документов в ФНС лично, по доверенности или почтовым курьером;
- получение подтверждения в течение 5 рабочих дней.
Если у индивидуального предпринимателя были сотрудники, то дополнительно необходимо будет решать вопросы с выплатой заработной платы, оформлением документации в связи с увольнением наемного персонала.
В случае, когда гражданин прекратил свою деятельность добровольно и не имеет долгов, то открыть закрытое ИП не составит труда. Законом не предусмотрены ограничения для проведения подобных действий. Гражданин вправе в любое время подать заявление в ФНС по месту жительства на оформление предпринимательской деятельности. Но в этом случае следует учесть, что восстановить старые данные уже не получится. Прежний правовой статус утратил свою силу, поэтому необходимо заново пройти процедуру регистрации бизнеса. Если направление работы бизнесмена и вид его деятельности изменились, то это обстоятельство требуется учесть при подаче заявления по форме Р21001.
Законом не предусмотрены ограничения для проведения подобных действий. Гражданин вправе в любое время подать заявление в ФНС по месту жительства на оформление предпринимательской деятельности. Но в этом случае следует учесть, что восстановить старые данные уже не получится. Прежний правовой статус утратил свою силу, поэтому необходимо заново пройти процедуру регистрации бизнеса. Если направление работы бизнесмена и вид его деятельности изменились, то это обстоятельство требуется учесть при подаче заявления по форме Р21001.
Если закрытие ИП произошло по решению суда и у гражданина множество долгов, то прежде чем открыть новый бизнес, необходимо позаботиться об устранении прошлых последствий. Следует заметить, что срок исковой давности составляет 3 года. В течение этого времени предпринимателю требуется погасить долговые обязательства. Только после этого можно подавать заявление на открытие нового ИП с другими реквизитами.
Сроки восстановления предпринимательства после ликвидации
В случае, когда ИП закрыла налоговая служба по банкротству, то требуется подождать 5 лет до открытия нового бизнеса. Срок устанавливается с момента прекращения судебных разбирательств по ликвидации юридического статуса компании.
Сотрудники ФНС ведут учет подобных случаев, поэтому не получится обмануть и зарегистрироваться раньше. В налоговую службу отправляется копия решения суда, которая фиксируется в документах.
Есть ли преимущество при повторной регистрации?
Многие бывшие предприниматели ошибочно полагают, что для восстановления статуса ИП после его закрытия для них будет применен упрощенный порядок. На самом деле процедура открытия и регистрации бизнеса во второй и последующий раз ничем не отличается от первичного процесса.
По-прежнему необходимо выполнить следующие шаги:
- подобрать коды ОКВЭД, соответствующие видам деятельности гражданина;
- заполнить заявление, подготовить пакет документов: паспорт, сведения ИНН, чек, подтверждающий оплату госпошлины;
- оформить нотариальную доверенность, в случае подачи бумаг в ФНС через представителя;
- оплатить обязательный сбор в госбюджет;
- подать соответствующее прошение о применении выбранной системы налогообложения;
- направить собранные документы в налоговую инспекцию.

После того как специалист ФНС проверит полноту данных, он выдаст расписку с указанием даты изготовления нового свидетельства о регистрации бизнеса.
По факту открытия ИП гражданин имеет право оформлять расчетный счет в банке, подключать онлайн-кассу, подавать заявление на установку эквайринга. Крупные кредитно-финансовые учреждения предлагают полный комплект услуг для индивидуальных предпринимателей: от регистрации бизнеса до подключения онлайн-бухгалтерии. Так, для клиентов Локо-Банка разработаны тарифные планы, позволяющие выбирать стоимость обслуживания РКО в зависимости от внешних переводов. Регистрация бизнеса происходит на бесплатной основе с полным оформлением необходимых документов. Такой подход исключает риск ошибок и отказа в получении правого статуса ИП.
Таким образом, закрыть и снова повторно открыть бизнес предприниматель может без ограничений. Все зависит от того, на каком основании происходило прекращение деятельности. Если гражданин не совершал правонарушений, то для него нет препятствий восстановления статуса ИП. В противном случае потребуется устранять последствия предыдущей деятельности перед подачей заявления на получения свидетельства о регистрации бизнеса.
В противном случае потребуется устранять последствия предыдущей деятельности перед подачей заявления на получения свидетельства о регистрации бизнеса.
Отказ в закрытии ИП: причины, устранение проблем
Если вы хотите прекратить предпринимательскую деятельность, вполне вероятно получить отказ в закрытии ИП. Рассмотрим в настоящей статье, как надлежащим образом подготовить документы и подать их в территориальное отделение ФНС, где факт закрытия ИП будет официально зарегистрирован с внесением записи о данном изменении в ЕГРИП.
Причины
В целом ряде случаев (они указаны в п. 1 ст. 23 Федерального закона № 129) органы ФНС могут выдать отказ. Каким бы ни было основание для отказа, ФНС обязана приложить документ, подробно описывающий нарушения, послужившие причиной отказа. Ниже мы детально рассмотрим встречающиеся случаи.
Каким бы ни было основание для отказа, ФНС обязана приложить документ, подробно описывающий причины или нарушения.
Прежде чем начинать процедуру, ознакомьтесь с пошаговой инструкцией, разработанной самими налоговиками. Она представлена здесь.
Она представлена здесь.
Распространенные основания для отказа:
- Представление документов на закрытие ИП не в тот территориальный орган. Помните, подачу документов на закрытие можно производить только в орган по месту государственной регистрации ИП.
- Ошибка нотариуса. Нотариусы – тоже люди и могут ошибаться. Если нотариус при заверении документов допустил техническую ошибку – это также послужит причиной для отказа в регистрации.
- Неправильное заполнение формы заявления на закрытие ИП в части указания паспортных данных. Данные вашего паспорта должны полностью совпадать с теми, что указаны в заявлении.
- Непредставление документов. Для закрытия ИП требуется представить полный пакет документов. Если вы забудете, скажем, ксерокопию паспорта, также получите отказ.
- Неправильная подпись на заявлении. Заявление на закрытие ИП может быть подписано только самим индивидуальным предпринимателем. Подписание заявления кем-либо еще повлечет отказ органов ФНС в регистрации.
 Обратите внимание, что подпись должна быть идентична подписи в паспорте.
Обратите внимание, что подпись должна быть идентична подписи в паспорте. - Наличие судебного запрета на регистрационные действия (как правило, это связано с возбуждением административного производства в службе судебных приставов).
- Нарушения со стороны индивидуального предпринимателя требований о порядке предоставления информации в Пенсионный фонд.
Если в налоговую направляется информация из судебных органов или от службы судебных приставов о том, что для данного ИП вводится запрет на осуществление регистрационных действий, связанных с прекращением им предпринимательской деятельности, то это обязательно должно делаться до внесения записи о регистрации ликвидации.
Основная проблема
Самой частой причиной отказа в регистрации служит наличие задолженности перед государственными фондами. Имейте в виду, что при ликвидации ИП его долги автоматически ложатся на физическое лицо, бывшее ранее предпринимателем. Но у предпринимателя, которому отказали на таком основании, есть шансы выиграть дело в случае обращения в суд. Даже при наличии серьезных задолженностей прохождение в суде процедуры банкротства позволяет ликвидировать ИП.
Даже при наличии серьезных задолженностей прохождение в суде процедуры банкротства позволяет ликвидировать ИП.
Самой частой причиной отказа в регистрации служит наличие задолженности.
Решение налогового органа, отказавшего в проведении регистрационных действий, должно быть оформлено письменно, чтобы в дальнейшем его можно было прикрепить к материалам дела. Исключением является только задолженность перед Пенсионным фондом. Отказ в регистрации при наличии подобной задолженности является правомерным.
границ | Роль структуры в биологии передачи сигналов интерферона
Введение
IFN были открыты более 60 лет назад (1957) как вещества, защищающие клетки от вирусной инфекции (1, 2). На основании их чувствительности к pH IFN были обозначены как тип I (pH-стабильный) или тип-II (pH-чувствительный) (2, 3). Характеристика их различных аминокислотных последовательностей и кристаллических структур (4, 5) (6-8) дополнительно подтвердила классификацию IFNα / β и IFNγ как IFN типа I и типа II, соответственно. Семейство типа I расширилось (9) и включило 12 IFNα (10–13), кодируемых 13 генами (IFNα1 / 13 кодирует тот же белок), IFNβ, IFNϵ (14), IFNκ (15) и IFNω (16). Геномный анализ в 2003 г. выявил новое семейство IFN типа III (IFNλ) (17, 18), которое с помощью анализа последовательности и последующего структурного анализа (19) было сходным с цитокинами семейства IL10 (12, 20–22), в частности, с IL-22. (23, 24). С открытием IFNλ4 в 2013 г. (25) в общей сложности 21 IFN (таблица 1) проявляет не только противовирусную активность, но и противоопухолевое действие, а также способность модулировать адаптивный иммунный ответ.
Семейство типа I расширилось (9) и включило 12 IFNα (10–13), кодируемых 13 генами (IFNα1 / 13 кодирует тот же белок), IFNβ, IFNϵ (14), IFNκ (15) и IFNω (16). Геномный анализ в 2003 г. выявил новое семейство IFN типа III (IFNλ) (17, 18), которое с помощью анализа последовательности и последующего структурного анализа (19) было сходным с цитокинами семейства IL10 (12, 20–22), в частности, с IL-22. (23, 24). С открытием IFNλ4 в 2013 г. (25) в общей сложности 21 IFN (таблица 1) проявляет не только противовирусную активность, но и противоопухолевое действие, а также способность модулировать адаптивный иммунный ответ.
Таблица 1 Семейства IFN и их рецепторные комплексы.
Плеотропная биологическая активность трех семейств IFN инициируется связыванием и последующей сборкой гетеродимерных рецепторных комплексов на клеточной мембране (таблица 1). 16 IFN типа I связываются и передают сигнал через рецепторный комплекс IFNAR1 и IFNAR2, IFNγ типа II связывается с цепями IFNGR1 и IFNGR2, а IFN типа III передает сигнал через рецепторные цепи IFNλR1 и IL-10R2. Каждый рецепторный гетеродимер состоит из рецепторной цепи с высоким сродством (например,g., IFNAR2, IFNGR1, IFNλR1) и рецепторной цепи с низким сродством к IFN (IFNAR1, IFNGR2, IL10R2). Рецепторы с высоким и низким сродством проявляют сродство в нМ и мкМ / мМ, соответственно, к своим родственным IFNs (26–30). Несмотря на различную аффинность, рецепторы типа I и типа II с высоким и низким сродством специфичны для родственных им членов семейства IFN. Напротив, IFNλR1 специфичен для членов семейства IFNλ типа III, но цепь IL-10R2 с низким сродством является общим рецептором, который также участвует в сигнальных комплексах IL10, IL22 и IL26 (12, 31–33).
Каждый рецепторный гетеродимер состоит из рецепторной цепи с высоким сродством (например,g., IFNAR2, IFNGR1, IFNλR1) и рецепторной цепи с низким сродством к IFN (IFNAR1, IFNGR2, IL10R2). Рецепторы с высоким и низким сродством проявляют сродство в нМ и мкМ / мМ, соответственно, к своим родственным IFNs (26–30). Несмотря на различную аффинность, рецепторы типа I и типа II с высоким и низким сродством специфичны для родственных им членов семейства IFN. Напротив, IFNλR1 специфичен для членов семейства IFNλ типа III, но цепь IL-10R2 с низким сродством является общим рецептором, который также участвует в сигнальных комплексах IL10, IL22 и IL26 (12, 31–33).
Образование рецепторного комплекса IFN активирует киназы Janus (JAK), которые инициируют IFN-опосредованные внутриклеточные сигнальные каскады (34–38). JAK конститутивно связываются с внутриклеточными доменами (ICD) рецепторов IFN посредством нековалентных взаимодействий (Таблица 1). Рецепторы IFN типа I и типа III используют одни и те же JAK для передачи сигнала. Рецепторы IFNAR2 и IFNλR1 с высоким сродством связываются с JAK1, тогда как рецепторы IFNAR1 и IL10R2 с низким сродством связываются с TYK2. Напротив, IFNGR1 и IFNGR2 типа II ассоциируют с JAK1 и JAK2, соответственно (39, 40).ICD рецепторов с низким сродством составляют 69–100 аминокислот, и их основная цель, по-видимому, состоит в связывании соответствующих киназ для активации при образовании рецепторного комплекса. ICD рецепторов с высоким сродством имеют длину от 223 до 271 аминокислот и содержат несколько остатков тирозина, которые при фосфорилировании JAK привлекают STAT, которые сами фосфорилируются, и перемещаются в ядро, где они активируют гены, стимулированные интерфероном (ISG) (40). , 41). Помимо использования одних и тех же JAK, IFN типа I и типа III индуцируют один и тот же транскрипционный комплекс STAT1 / STAT2 / IRF9, ISGF3 (40–42).IFNγ активирует гомодимеры фосфо-STAT1, но не ISGF3, что отражается в ~ 1000 раз более низкой противовирусной активности IFNγ по сравнению с IFN типа I и типа III (43, 44).
Рецепторы IFNAR2 и IFNλR1 с высоким сродством связываются с JAK1, тогда как рецепторы IFNAR1 и IL10R2 с низким сродством связываются с TYK2. Напротив, IFNGR1 и IFNGR2 типа II ассоциируют с JAK1 и JAK2, соответственно (39, 40).ICD рецепторов с низким сродством составляют 69–100 аминокислот, и их основная цель, по-видимому, состоит в связывании соответствующих киназ для активации при образовании рецепторного комплекса. ICD рецепторов с высоким сродством имеют длину от 223 до 271 аминокислот и содержат несколько остатков тирозина, которые при фосфорилировании JAK привлекают STAT, которые сами фосфорилируются, и перемещаются в ядро, где они активируют гены, стимулированные интерфероном (ISG) (40). , 41). Помимо использования одних и тех же JAK, IFN типа I и типа III индуцируют один и тот же транскрипционный комплекс STAT1 / STAT2 / IRF9, ISGF3 (40–42).IFNγ активирует гомодимеры фосфо-STAT1, но не ISGF3, что отражается в ~ 1000 раз более низкой противовирусной активности IFNγ по сравнению с IFN типа I и типа III (43, 44). Помимо активации отдельных внутриклеточных сигнальных путей, IFN типа I / III продуцируются в клетках при вирусной инфекции или инфекции другими патогенами через рецепторные пути распознавания образов, включая RIGI, MDA7, PKR, TLR3, TLR7, TLR9 и STING. (40, 45–48). Напротив, IFNγ типа II продуцируется преимущественно антиген-активированными Т-лимфоцитами (39).Таким образом, IFN типа I / III являются продуктами врожденной иммунной системы, разработанными для установления прямого и немедленного противовирусного состояния в клетках, но также могут модулировать адаптивные иммунные ответы. IFNγ типа II сам по себе является продуктом адаптивного иммунитета, который действует на клетки врожденного иммунитета, особенно на макрофаги. Как мощный активатор макрофагов, IFNγ необходим для борьбы с микобактериями и другими внутриклеточными патогенами (49, 50). Дефицит IFNGR1 у людей связан с микобактериальными инфекциями, в то время как люди с дефицитом IFNAR2 или IFNAR1 имели опасное для жизни заболевание после вакцинации вакцинами против эпидемического паротита, кори и краснухи (MMR) (51, 52).
Помимо активации отдельных внутриклеточных сигнальных путей, IFN типа I / III продуцируются в клетках при вирусной инфекции или инфекции другими патогенами через рецепторные пути распознавания образов, включая RIGI, MDA7, PKR, TLR3, TLR7, TLR9 и STING. (40, 45–48). Напротив, IFNγ типа II продуцируется преимущественно антиген-активированными Т-лимфоцитами (39).Таким образом, IFN типа I / III являются продуктами врожденной иммунной системы, разработанными для установления прямого и немедленного противовирусного состояния в клетках, но также могут модулировать адаптивные иммунные ответы. IFNγ типа II сам по себе является продуктом адаптивного иммунитета, который действует на клетки врожденного иммунитета, особенно на макрофаги. Как мощный активатор макрофагов, IFNγ необходим для борьбы с микобактериями и другими внутриклеточными патогенами (49, 50). Дефицит IFNGR1 у людей связан с микобактериальными инфекциями, в то время как люди с дефицитом IFNAR2 или IFNAR1 имели опасное для жизни заболевание после вакцинации вакцинами против эпидемического паротита, кори и краснухи (MMR) (51, 52). Вместе эти данные подчеркивают различные роли этих IFN в борьбе с различными патогенами.
Вместе эти данные подчеркивают различные роли этих IFN в борьбе с различными патогенами.
Хотя существует только один IFNγ, примечательно, что люди кодируют 16 различных IFN типа I и 4 IFN типа III, которые индуцируют одну и ту же фундаментальную ISGF3-опосредованную антивирусную программу в клетках (17, 18, 53, 54). Необходимость этого замечательного арсенала IFN для борьбы с вирусами и другими патогенами (55–58) остается областью интенсивных исследований. Учитывая сложность передачи сигналов IFN, этот обзор описывает фундаментальную структурную организацию каждого рецепторного комплекса IFN в генерировании ответов передачи сигналов IFN.Основное внимание уделяется определению того, как структура влияет на сродство к рецептору IFN-IFN, специфичность и роль общей архитектуры комплекса в позиционировании ICD рецептора для внутриклеточной активации JAK / STAT и последующей клеточной активности.
Структуры IFN типа I, типа II и типа III
Все IFN имеют α-спиральные структуры с уникальной топологией вверх-вверх-вниз-вниз (21) по сравнению с другими белками пучка α-спиралей ( Рисунок 1). Каждый IFN состоит из шести вторичных структурных элементов, обозначенных A-F, из которых спирали A, C, D и F образуют антипараллельный четырехспиральный пучок.Петлевые элементы B и E демонстрируют более вариабельные вторичные структуры, от дополнительных спиралей до протяженных сегментов, которые упаковываются по краю четырехспирального пучка (спирали A, C, D и F). Α-Спирали IFN типа I длинные, прямые и по существу параллельны друг другу (рис. 1A). Несмотря на значительное разнообразие последовательностей (35–95%), все 16 IFN имеют одинаковую α-спиральную структуру (4, 5, 59–63). В отличие от IFN типа I, IFN типа III состоят из более коротких спиралей, которые содержат несколько перегибов, которые образуют более компактный пучок (рис. 1B).В результате IFN типа III принимают структуры, которые больше похожи на цитокин IL-22 семейства IL-10, чем на IFN типа I (12, 19, 23, 24, 64). Это интересно с функциональной точки зрения, поскольку IL-22 индуцирует антибактериальную активность в кишечнике и коже через ограниченный тканью рецепторный комплекс IL22R1 и IL10R2 (22, 32, 65–70).
Каждый IFN состоит из шести вторичных структурных элементов, обозначенных A-F, из которых спирали A, C, D и F образуют антипараллельный четырехспиральный пучок.Петлевые элементы B и E демонстрируют более вариабельные вторичные структуры, от дополнительных спиралей до протяженных сегментов, которые упаковываются по краю четырехспирального пучка (спирали A, C, D и F). Α-Спирали IFN типа I длинные, прямые и по существу параллельны друг другу (рис. 1A). Несмотря на значительное разнообразие последовательностей (35–95%), все 16 IFN имеют одинаковую α-спиральную структуру (4, 5, 59–63). В отличие от IFN типа I, IFN типа III состоят из более коротких спиралей, которые содержат несколько перегибов, которые образуют более компактный пучок (рис. 1B).В результате IFN типа III принимают структуры, которые больше похожи на цитокин IL-22 семейства IL-10, чем на IFN типа I (12, 19, 23, 24, 64). Это интересно с функциональной точки зрения, поскольку IL-22 индуцирует антибактериальную активность в кишечнике и коже через ограниченный тканью рецепторный комплекс IL22R1 и IL10R2 (22, 32, 65–70). Таким образом, IFNλ и IL-22 контролируют вирусные и бактериальные проблемы, соответственно, на поверхности барьера (22, 64, 71). Как «интерфероны слизистой оболочки», IFNλ рекламируются как оптимальное лекарство для лечения респираторных вирусов, таких как коронавирус 2 тяжелого острого респираторного синдрома (SARS-CoV-2), вызывающий COVID-19 (72).Однако передача сигналов IFNλ у мышей предотвращает восстановление эпителия легких, что приводит к бактериальным суперинфекциям (73, 74). Другие исследования показывают, что IFN типа I, а не IFN-λ, могут быть наиболее эффективными и безопасными при лечении SARS-CoV-2 (75). В целом, эти исследования подчеркивают сложность передачи сигналов IFN на поверхностях барьеров и различия в результатах передачи сигналов IFN у мышей и людей.
Таким образом, IFNλ и IL-22 контролируют вирусные и бактериальные проблемы, соответственно, на поверхности барьера (22, 64, 71). Как «интерфероны слизистой оболочки», IFNλ рекламируются как оптимальное лекарство для лечения респираторных вирусов, таких как коронавирус 2 тяжелого острого респираторного синдрома (SARS-CoV-2), вызывающий COVID-19 (72).Однако передача сигналов IFNλ у мышей предотвращает восстановление эпителия легких, что приводит к бактериальным суперинфекциям (73, 74). Другие исследования показывают, что IFN типа I, а не IFN-λ, могут быть наиболее эффективными и безопасными при лечении SARS-CoV-2 (75). В целом, эти исследования подчеркивают сложность передачи сигналов IFN на поверхностях барьеров и различия в результатах передачи сигналов IFN у мышей и людей.
Рисунок 1 Структуры членов семейства IFN. Схема и ленточные диаграммы показывают шесть вторичных структурных элементов типа I (A) , pdbid = 1AU, типа III (B) , pdbid = 3HHC и типа II (C) , pdbid = 6E3K IFN. Структуры IFN окрашены в цвет радуги от N-концевой спирали A (синий) до C-концевой спирали F (красный).
Структуры IFN окрашены в цвет радуги от N-концевой спирали A (синий) до C-концевой спирали F (красный).
В отличие от мономерных IFN типа I и типа III, IFNγ принимает структуру интеркалированного димера, где спирали E и F из одной цепи «меняются местами» с другой субъединицей димера (рис. 1C). Как и IFNλ, структура IFNγ наиболее сходна с IL10, который является членом-основателем семейства цитокинов IL-10 (12, 21, 32, 76–78). Эти данные подтверждают, что каждое семейство IFN принимает отдельный α-спиральный каркас, который должен «обрабатывать» различные степени вариации последовательности, чтобы регулировать взаимодействие своих клеточных рецепторов.Например, существует один высококонсервативный димер IFNγ типа II, тогда как существует 16 мономерных IFN типа I (идентичность последовательностей 35–95%) и 4 IFN типа III (идентичность последовательностей 28–96%), которые проявляют вариабельность. идентичность аминокислотных последовательностей. Это подчеркивает различные механизмы, используемые каждым семейством IFN для регулирования биологической активности. Гомодимеризация рецептора с помощью IFNγ по сравнению с вариабельными контактами IFN / IFN-рецептора с помощью мономерных IFN типа I и типа III. Эти механизмы будут рассмотрены более подробно ниже.
Гомодимеризация рецептора с помощью IFNγ по сравнению с вариабельными контактами IFN / IFN-рецептора с помощью мономерных IFN типа I и типа III. Эти механизмы будут рассмотрены более подробно ниже.
Комплекс IFNλ / IFNλR1 / IL10R2 типа III
Рецепторный комплекс IFNλ типа III (79) демонстрирует простейшую архитектуру из трех семейств IFN. Мономерные IFNλ собирают сигнальные комплексы 1: 1: 1 с рецепторами IFNλR1 с высоким сродством и рецепторами IL10R2 с низким сродством (рис. 2A). IFNλR1 и IL10R2 оба состоят из двух β-сэндвич-доменов (D1, D2), где домены D2 расположены ближе всего к мембране. IFNλR1 связывается с IFNλ с помощью пяти рецепторных петель (L2-L6), которые расположены на стыке доменов D1 и D2.Петли связывания IFNλR1 контактируют с остатками IFNλ, расположенными на спирали A, петле AB и спирали F. Несмотря на различия в деталях, сайт связывания сайта-1 с высоким сродством IFNλ / IFNλR1 сохраняется с рецепторными комплексами с высоким сродством типа I и типа II. (Фигура 2). Низкоаффинный сайт связывания IL10R2-2 состоит из N-концевых остатков IFNλ до начала спирали A (например, область пре-A (80), также см. Рисунок 3A), остатков на спирали C и в сегменте спирали D, которая проходит параллельно области pre-A.IL10R2 использует подмножество тех же петель, используемых IFNλR1 (петли L2, L3 и L5) для связи с IFNλ. Таким образом, интерфейс IFNλ-IL10R2 сайт-2 является прерывистым, создавая меньший контакт L2 / спираль D (сайт-2a) и большее взаимодействие между L3 / L5 и пре-A IFNλ и спиралью D (сайт 2b).
(Фигура 2). Низкоаффинный сайт связывания IL10R2-2 состоит из N-концевых остатков IFNλ до начала спирали A (например, область пре-A (80), также см. Рисунок 3A), остатков на спирали C и в сегменте спирали D, которая проходит параллельно области pre-A.IL10R2 использует подмножество тех же петель, используемых IFNλR1 (петли L2, L3 и L5) для связи с IFNλ. Таким образом, интерфейс IFNλ-IL10R2 сайт-2 является прерывистым, создавая меньший контакт L2 / спираль D (сайт-2a) и большее взаимодействие между L3 / L5 и пре-A IFNλ и спиралью D (сайт 2b).
Рисунок 2 Структуры рецепторных комплексов IFN. Ленточные диаграммы типа III (A) , pdbid = 5T5W, типа II (B) , pdbid = 6E3K и типа I (C) , pdbid = 3SE4, рецепторные комплексы.IFN окрашены в цвет радуги, как показано на рисунке 1. β-тяжи цепей рецепторов с высоким сродством окрашены в зеленый цвет, а цепи с низким сродством — в пурпурный. Показано, что для рецепторного комплекса IFNγ типа II только одна субъединица IFNγ подчеркивает сходство «половины» комплекса с рецепторным комплексом IFN-типа III. Разделение С-концов рецепторных цепей IFNλR1 / IL-10R2 типа III и IFNGR1 / IFNGR2 типа II, где они входят в мембрану, составляет 30 Å и 22 Å, соответственно. Взаимодействие D2-D4 не наблюдалось в структурах комплекса IFN / IFNAR1 / IFNAR2.
Разделение С-концов рецепторных цепей IFNλR1 / IL-10R2 типа III и IFNGR1 / IFNGR2 типа II, где они входят в мембрану, составляет 30 Å и 22 Å, соответственно. Взаимодействие D2-D4 не наблюдалось в структурах комплекса IFN / IFNAR1 / IFNAR2.
Рисунок 3 Незначительные структурные изменения между IFNλ1 / IFNλ3 изменяют контакты IFNλR1. (A) Альфа-углеродная диаграмма суперпозиции IFNλ1 и IFNλ3. Расположение структурных различий в областях петли B IFNλ1 и IFNλ3, как обсуждается в тексте, обведено кружком. (B) Увеличение «пролинового переворота» петли B, наблюдаемое в структурах IFNλ1 и IFNλ3, и его влияние на конформацию Arg-180 IFNλ3 (зеленый), где он образует солевой мостик с IFNλR1 Asp-91.Напротив, IFNλ1 Arg-175 (пурпурный) простирается от IFNλR1 Asp-91 к петле B.
В дополнение к контактам IFNλ-IL10R2 site-2, IL10R2 формирует дополнительный интерфейс D2-D2 site-3 с IFNλR1. Таким образом, полный сайт связывания IL10R2 образуется только после того, как IFNλ связывается с IFNλR1. Эта структурная организация обеспечивает кооперативное образование рецепторного комплекса IFNλ, при котором сначала формируется комплекс IFNλ / IFNλR1, а затем происходит связывание IL10R2 с сайтом-2 и сайтом-3. После образования собранный комплекс IFNλ позиционирует С-концевые концы IFNλR1 и IL10R2 на расстоянии 30 Å друг от друга перед входом в мембрану.Комбинированные интерфейсы сайт-2 и сайт-3 скрывают более 1500 Å (2) площади поверхности, что более чем в два раза превышает площадь поверхности, захороненную во взаимодействии с высоким сродством IFNλ3 / IFNλR1 сайт-1. Однако, несмотря на такой обширный интерфейс, энергетически критических взаимодействий мало. Таким образом, сродство IL-10R2 к комплексу IFNλ3 / IFNλR1 (например, сайт-2 + сайт-3) составляет 15 мкМ (79), что примерно в 15 раз ниже, чем сродство IFNAR1 к большинству подтипов IFN (26, 27). В то время как IFNλ3 / IFNλR1 представляет собой взаимодействие с «высоким сродством» в комплексе, измеренное значение K D для 850 нМ (79) примерно на 1 log ниже, чем сродство самого слабого IFN типа I к IFNAR2 (например.g., IFNα1, K D ~ 100 нМ).
Из-за низкого сродства IFNλ к их рецепторам, IFNλ чувствительны к уровням экспрессии их рецепторов на клетках. Фактически, главное различие между ИФН типа I и типа III заключается в уникальном распределении их рецепторов на разных типах клеток (81, 82). Рецепторы IFNAR1 и IFNAR2 типа I присутствуют на всех ядросодержащих клетках, в то время как экспрессия IFNλR1 преимущественно ограничивается эпителиальными клетками, как упоминалось ранее для IL22R1 (22, 70).Таким образом, передача сигналов IFNλ, по-видимому, специализируется на борьбе с вирусными инфекциями на поверхностях эпителиального барьера, таких как легкие, кишечник и печень (83). Это наиболее впечатляюще было продемонстрировано путем демонстрации того, что IFNλ, но не IFN типа I, важен для борьбы с норовирусной инфекцией (84). Хотя эпителиальные клетки кишечника в этом исследовании экспрессируют IFNAR типа I, их экспрессия ограничена апикальной поверхностью клеток, а на базолатеральной поверхности экспрессии IFNAR не наблюдается. Таким образом, избирательная передача сигналов IFN-λ в эпителиальных клетках кишечника была полностью оценена только в рамках организации интактного кишечника у животных.В то время как активность IFNλ кажется «слабой» во многих клеточных анализах, данные in vivo и предполагают сильную передачу сигналов IFNλ в контексте тканей и органов. Следует отметить, что IFN типа I, IFNϵ и IFNκ, защищают женский репродуктивный путь (85–87) и кожу (15, 88) соответственно. Примечательно, что, как и IFNλ, IFNϵ и IFNκ проявляют «низкое» сродство к рецепторам типа I по сравнению с большинством IFN типа I (89).
Выводы из двоичных структур IFNλ1 / IFNλR1 и IFNλ3 / IFNλR1
Были решены бинарные комплексные структуры как IFNλ1 / IFNλR1, так и IFNλ3 / IFNλR1 (79, 90).IFNλ1 и IFNλ3 имеют очень похожие структуры со среднеквадратичным отклонением (среднеквадратичное отклонение) 0,6 Å. Точно так же связывание IFNλR1 с IFNλ1 или IFNλ3 демонстрирует среднеквадратичное значение. 0,68 Å. Наконец, структура несвязанного IL10R2 (91) и IL10R2, связанного с IFNλ3, демонстрирует среднеквадратичное значение. 1,3Å. Чем больше среднеквадратичное значение происходит из-за изменений конформации петли связывания IL10R2 L5 при контакте с IFNλ3. Несмотря на это различие, общие структуры связанного и несвязанного IL10R2 одинаковы. Эти структурные сравнения предполагают, что все IFNλs собирают сигнальный комплекс с одинаковой общей архитектурой.Таким образом, биологическая активность IFNλ регулируется не структурой тройного комплекса, а сродством каждого IFNλ к цепям IFNλR1 и IL10R2 и, в конечном итоге, стабильностью комплекса.
Клеточные анализы in vitro демонстрируют, что IFNλ3 проявляет в два раза большую противовирусную активность, чем IFNλ1 (92). Хотя подробный анализ аффинности связывания рецептора IFNλ не был завершен, мы ожидаем, что комплекс IFNλ3 / IFNλR1 должен демонстрировать отличия от комплекса IFNλ1 / IFNλR1, что согласуется с более высоким взаимодействием сродства.Сравнение структур IFNλ1 и IFNλ3 (рис. 3A) показывает, что участки петли B IFNλ1 и IFNλ3 демонстрируют разные конформации, в частности, Pro-74 IFNλ1 / Pro-77 IFNλ3 (рис. 3B). В IFNλ3 Pro-77 движется к спирали F, в то время как в IFNλ1 Pro-74 движется от спирали F. Этот «переворот пролина» изменяет положение консервативного Arg-175 IFNλ1 / Arg-180 IFNλ3 , расположенного на спирали F (рис. 3В). В IFNλ3 гуанидиногруппа Arg-180 упаковывается против Pro-77, который позиционирует ее для двухвалентного солевого мостика с остатком IFNλR1 Asp-91.Серия мутантов IFNλ3 по аланину была протестирована на противовирусную активность и идентифицировала Phe-179 как наиболее важный остаток IFNλ3 для индукции противовирусной активности (19). Поскольку IFNλ3 Phe-179 соседствует с Arg-180, вполне вероятно, что мутация Phe-179 в аланин разрушает солевой мостик Arg-180 IFNλ3 / Asp-91 IFNλR1 , что снижает сродство связывания IFNλR1 и противовирусную активность. .
«переворот пролина», наблюдаемый между IFNλ1 и IFNλ3 (рис. 3B), также может дать механистическое понимание сниженной биологической активности однонуклеотидного полиморфизма (SNP) IFNλ4, rs117648444.Rs11768444 соответствует IFNλ4-Pro70Ser, который проявляет пониженную противовирусную активность по сравнению с IFNλ4 дикого типа (25, 93). Понимание SNP IFNλ4 важно, поскольку несколько групп сопоставили основную генетическую детерминанту клиренса вируса гепатита C (HCV) в ответ на лечение IFN-α плюс рибавирин с локусами IFN типа III (94–96). В конечном счете, активность IFNλ4 была вовлечена в качестве причинного фактора нарушения клиренса HCV у пациентов, которые кодируют «активный» белок IFNλ4, в отличие от неактивного белка IFNλ4 (25).Несмотря на общую ~ 28% идентичность последовательности с IFNλ3, IFNλ4 принимает ту же α-спиральную складку, что и другие IFNλ, и связывается с IFNλR1 и IL10R2 (97). Выравнивания аминокислотных последовательностей показывают, что IFNλ4 Pro-70 идентичен IFNλ3 Pro-77, предполагая, что мутация IFNλ4 Pro70Ser влияет на взаимодействия IFNλ4-IFNλR1, изменяя структуру IFNλ4 Arg-163, как описано для Arg-180 в IFNλ3 (рис. 3B).
IFNλ2 не был изучен в той же степени, что и другие IFNλ, предположительно потому, что было показано, что он проявляет противовирусную активность в ~ 5–10 раз ниже (53, 98).Аминокислотная последовательность IFNλ2 отличается от IFNλ3 всего на 6 аминокислот. Моделирование структуры IFNλ2 на основе структуры IFNλ3 предполагает, что R28H находится в неструктурированной области на N-конце молекулы, где не ожидается, что он изменяет связывание с рецептором. K70R и R72H расположены в петле AB IFNλ2, но не контактируют с IFNλR1. Кроме того, мутант IFNλ3 R72A снижал противовирусную активность IFNλ3 только на 30%, что позволяет предположить, что эти изменения остатков не могут объяснить более низкую активность IFNλ2.Остатки V92M и h256Y расположены на открытых поверхностях спиралей C и E IFNλ2, соответственно, которые расположены напротив сайтов связывания IFNλR1 и IL10R2. Таким образом, если бы эти аминокислоты были ответственны за более низкую активность IFNλ2, это поддерживало бы гипотезу о некоторых группах, которые IFNλ могут связываться с другой, неидентифицированной рецепторной цепью (83). Наконец, L133F расположен на спирали D, где боковая цепь скрыта в гидрофобном ядре IFNλ2. Замена аминокислот с L на F не может быть включена в гидрофобное ядро структуры IFNλ3 без искажения спиралей A, D или F.Это предполагает, что L133F может быть основным остатком, ответственным за снижение биологической активности IFNλ2 по сравнению с IFNλ3.
Комплекс IFNγ / IFNGR1 / IFNGR2 типа II
Рецепторный комплекс IFNγ типа II обеспечивает важную структуру для дальнейшего понимания комплексов типа I и типа III (99). Уникальная структура интеркалированного димера (6) IFNγ отличает его от мономерных IFN типа I и типа III с дисульфидной связью (4, 19, 100). Димер IFNγ собирает симметричный гетеродимерный комплекс 1: 2: 2 IFNGR1 / IFNGR2 (99, 101) (рис. 4) по сравнению с гетеродимерными комплексами 1: 1: 1 IFN типа I и типа III (рис. 2). .В димерном комплексе двойные С-концы гетеродимеров IFNGR1 / IFNGR2 расположены на расстоянии 85 Å друг от друга. Как было предположено на основе анализа структурно родственного димера IL10 (102), димерный IFNγ позиционирует IFNGR1 и IFNGR2 (рисунок 4) и их соответствующие ICD в оптимальном димерном расположении для привлечения неактивных димеров STAT1 (103) для последующего фосфорилирования и активации. гомодимеров STAT1 (104). Нарушение архитектуры комплекса димерного рецептора IFNγ с использованием сконструированных мономерных IFNγ, которые собирают 1/2 димерного IFNγ / IFNG1 / IFNGR2 (см. Рис. 2 vs.Рисунок 4), резко снизили некоторые биологические активности, индуцированные IFNγ (7, 8, 99, 102, 105). Дополнительные мутанты IFNγ подтвердили, что димерное расположение IFNGR1, а не IFNGR2, важно для полного фосфорилирования STAT1 (99). В отличие от STAT1, многие дополнительные пути, активируемые IFNγ, включая киназу MAP, PI3K и CaMKII (106), по-видимому, не одинаково чувствительны к IFNγ-опосредованной димеризации IFNGR1 / IFNGR2. Таким образом, по крайней мере на некоторых клетках сконструированные мономеры IFNγ могут индуцировать такие же уровни HLA-A на клеточной поверхности, что и димер IFNγ дикого типа (99).Интересно отметить, что нейроны, по-видимому, естественным образом манипулируют результатами передачи сигналов IFNγ, поддерживая низкие уровни STAT1, что приводит к мощной IFNγ-опосредованной активации ERK1 / 2 (107). В целом, димерная архитектура комплекса IFNγ / IFNGR1 / IFNGR2 имеет решающее значение для индукции полного спектра плеотропной активности, опосредованной IFNγ (108), включая активацию макрофагов (109, 110), наблюдение за опухолью (111, 112) и защиту от внутриклеточных патогенов, в том числе микобактерий (50, 113).
Рисунок 4 Димерный комплекс IFNγ / IFNGR1 / IFNGR2. Ленточная диаграмма комплекса димер IFNγ 1: 2: 2 / IFNGR1 / IFNGR2 (pdbid = 6E3K). Показаны два вида на комплекс. Первая приблизительно перпендикулярна оси второго порядка IFNγ (A) , а вторая параллельна оси второго порядка (B) .
Несмотря на более крупную димерную сборку, внутри одной субъединицы IFNγ IFNGR1 и IFNGR2 образуют аналогичные интерфейсы сайта 1, сайта 2 и D2-D2 сайта 3, как ранее описано для комплекса IFNλ / IFNλR1 / IL10R2 (рис. 2B). .По сравнению с IFNλ / IFNλR1 интерфейс IFNγ site-1 более обширен с основными контактами между петлей AB и спиралью F петель IFNγ и IFNGR1 L2-L6. Интерфейс IFNγ / IFNGR2 сайта 2 состоит почти исключительно из контактов со спиралью D IFNγ и не имеет контактов со спиралью A, основной контактной областью в комплексе IFNλ. Несмотря на эти различия, IFNGR2 по-прежнему образует интерфейс сайта 3 D2-D2 с IFNGR1, который позиционирует C-концы рецепторов на расстоянии 22Å друг от друга на поверхности клетки до их входа в мембрану.Таким образом, сборка сигнального комплекса IFNγ является кооперативной, требующей сначала образования бинарного комплекса IFNγ / IFNGR1, а затем связывания IFNGR2 для индукции клеточной сигнализации.
Комплекс IFN / IFNAR1 / IFNAR2 типа I
Рецепторный комплекс IFN типа I отличается от рецепторных комплексов как типа II, так и типа III (рис. 2). Цепь IFNAR2 с высоким сродством принимает структуру рецептора D1 / D2 с двумя доменами, как это наблюдается для цепей IFNλR1 и IFNGR1 (рис. 2) (114). Структуры ЯМР и рентгеновские лучи подтверждают, что IFNAR2 связывается с эпитопом IFN сайта-1, который состоит из остатков спирали A, петли AB и спирали F, аналогично IFN типа II и типа III (100, 115, 116 ).IFNAR2 осуществляет обширные взаимодействия с Arg-33 (нумерация IFNα2) в петле AB IFN. Arg-33 и соседний по структуре Leu-30 составляют примерно две трети энергии связи IFNα2 / IFNAR2 (29, 100, 117). Дополнительные критические контакты происходят с петлями связывания L3 и L4 IFNAR2, которые контактируют с остатками F спирали Met-148 и Arg-149 (числа IFNα2) (117). Хотя нам известно, что все 16 IFN проявляют различную аффинность к IFNAR2 (26–28, 89), механизмы, которые контролируют сродство IFNAR2 к каждому подтипу IFN, остаются неполными.В общем, оказывается, что тонкие изменения остатков вокруг этих энергетически важных остатков модулируют аффинность IFN-подтипа IFNAR2.
Цепь низкоаффинного рецептора IFN типа I, IFNAR1, полностью уникальна по сравнению с другими цитокиновыми рецепторами семейства IFN и IL10 (рис. 2). IFNAR1 состоит из четырех β-сэндвич-доменов (D1-D4), подобных тандемным рецепторам D1 / D2, где домен D4 является проксимальным доменом мембраны. Домены D2 и D3 рецептора образуют обширный интерфейс друг с другом, в то время как домен D1 может подвергаться движениям твердого тела.В целом, домены IFNAR1 D1-D3 образуют IFN-связывающий модуль, в то время как домен D4 присоединяется к D3 с помощью гибкого линкера, который позволяет домену D4 принимать множественные конформации, даже когда он связан с IFN (100, 118). Несмотря на уникальную структуру, петли IFNAR1 на концах доменов D1, D2 и D3 контактируют со спиралями IFN C, D и E, при этом домен D1 «закрывается» на спирали E, как рука, схватившая стакан.
Основываясь на особенностях, описанных выше, связывание IFN типа I IFNAR1 представляет собой новую парадигму распознавания белков.Во-первых, поверхность контакта IFNAR1-IFN, состоящая из спиралей C, D и E IFN, больше, чем для других комплексов IFN. Во-вторых, проксимальный мембранный домен D4 IFNAR1 не образует интерфейс сайта 3, по крайней мере, не стабильный интерфейс с доменом D2 IFNAR2. Это говорит о том, что за счет увеличения размера интерфейса IFNAR1-IFN сайт-2 (см. Фиг. 2C), используя новые взаимодействия D1 / спираль E, комплекс IFN типа I больше не требует интерфейса сайта-3. Таким образом, для комплекса IFN типа I отсутствует кооперативность на основе структуры, обеспечиваемая взаимодействием сайта-3 D2-D4.Скорее, сборка и стабильность рецепторного комплекса полностью контролируются аффинностями IFN-IFNAR2 и IFN-IFNAR1. Хотя возможно, что свободные IFN и IFN, связанные с IFNAR2, могут проявлять различное сродство к IFNAR1, что приводит к основанному на аффинности механизму кооперативного связывания, это не было продемонстрировано экспериментально.
Механистическая роль домена IFNAR1 D4 в активации рецептора IFN типа I остается неясной, поскольку домен D4 не наблюдался в кристаллических структурах комплекса IFN / IFNAR1 / IFNAR2 (рис. 5A).Для определения возможного местоположения домена IFNAR1 D4 комплекс IFNλ3 / IFNλR1 / IL10R2 был наложен на комплекс IFNω / IFNAR1 / IFNAR2 (рис. 5B). В этой модели домен D1 IL10R2 перекрывается с доменом IFNAR1 D3, а предполагаемое местоположение домена IFNAR1 D4, представленного доменом IL10R2 D2, соседствует с доменом IFNAR2 D2, создавая интерфейс сайта-3 D2-D4, поскольку наблюдается в комплексах типа II и типа III (рис. 2). Второе возможное положение домена D4 обеспечивается структурой бинарного комплекса мышиного IFNβ / IFNAR1 (119), где наблюдались все четыре домена IFNAR1.Суперпозиция мышиного комплекса IFNβ / IFNAR1 на человеческий комплекс IFN / IFNAR1 / IFNAR2 размещает С-концевые концы IFNAR2 D2 и IFNAR1 D4 на расстоянии 51Å (рис. 5C), в отличие от 30Å и 22Å для комплексов IFNλ и IFNγ, соответственно. . Эти модели позволяют сделать два возможных вывода. Во-первых, IFN типа I собирают новый «открытый» комплекс с С-концевыми концами IFNAR1 и IFNAR2, разделенными на ~ 50 Å. Во-вторых, «открытая» конформация представляет собой неактивный комплекс, который должен «закрываться», чтобы образовать интерфейс сайта-3 D2 / D4 для индукции активности IFN.Наш анализ показывает, что связывание IFN с IFNAR2 и IFNAR1 способствует временным взаимодействиям IFNAR2-D2 / IFNAR1-D4. Таким образом, стабильность взаимодействия IFN / IFNAR1 / IFNAR2 будет контролировать количество временных «открытых» / «закрытых» событий связывания сайта-3 D2-D4, которые могут влиять на силу передачи сигналов. Таким образом, стабильность взаимодействий IFN / IFNAR2 и IFN / IFNAR1 будет регулировать передачу сигналов, как было описано ранее (120).
Рис. 5 Структурные модели домена IFNAR1 D4. (A) Ленточная диаграмма комплексной структуры типа I IFN (IFNω, синий) / IFNAR1 (оранжевый) / IFNAR2 (желтый) (pdbid = 3SE4), в которой отсутствует домен IFNAR1 D4. (B) Суперпозиция тройного комплекса IFNλ3 (радуга) / IFNλR1 (зеленый) / IL10R2 (пурпурный) на структуре IFN / IFNAR1 / IFNAR2 позиционирует домен IL10R2 D2 (пурпурный), так что он может представлять временное местоположение домен IFNAR1 D4, образующий стержневое взаимодействие IFNAR2 D2-IFNAR1 D4. (C) Второе возможное местоположение домена D4 IFNAR1 человека показано наложением комплекса IFNβ / IFNAR1 мыши (pdbid = 3WCY) на комплекс IFN / IFNAR1 / IFNAR2.Положение смоделированного домена D4 (зеленый), полученного из структуры мышиного IFNβ / IFNAR1, показано зеленым, а положение домена IFNAR1 D4, полученного в результате наложения рецепторного комплекса IFNλ, показано пурпурным. Поскольку домен D4 человеческого IFNAR1 не образует стабильного взаимодействия D2-D4 с IFNAR2, D4 может переходить между зеленой и пурпурной конформациями, чтобы вызвать биологическую активность. Точная роль домена D4 в передаче сигнала IFN остается неизвестной.
Несмотря на структуры, которые обнаруживают механизмы распознавания и сборки внеклеточного рецептора IFN, остаются вопросы о событиях передачи сигнала, опосредованных IFN, которые инициируют и поддерживают клеточную активацию.Например, остается неясным, каким образом все 16 IFN, которые проявляют спектр аффинности к IFNAR (слабый / сильный), могут все активировать подмножество генов, связанных с противовирусной активностью на всех клетках, в то время как дополнительные клеточные функции IFN, один такое считывание, являющееся антипролиферативной активностью, коррелирует с аффинностью IFN-IFNAR (121). Эти два различных клеточных считывания, обозначенные как надежная и настраиваемая активация (121), могут быть объяснены моделью предварительной ассоциации IFNAR1 / 2 (122) и моделью гетеродимеризации IFNAR1 / 2, опосредованной IFN (123), соответственно.Предварительная ассоциация IFNAR может объяснить быструю IFN-опосредованную активацию экспрессии противовирусного гена, тогда как IFN-опосредованная димеризация IFNAR может объяснить настраиваемую экспрессию гена. Смысл модели до ассоциации состоит в том, что IFN вызывают структурные изменения в IFNAR, которые активируют JAK1 / TYK2 и вызывают быструю экспрессию антивирусных генов, в то время как модель димеризации полагается исключительно на IFN-опосредованную димеризацию IFNAR для активации JAK1. / TYK2 и впоследствии индуцируют IFN-опосредованную экспрессию гена.Было высказано предположение, что технические вопросы, в частности, анализ искусственно высоких уровней экспрессии IFNAR, ответственны за наблюдение за предварительно ассоциированными IFNAR (123). К сожалению, исследователи, критикующие преассоциативную модель, не подтвердили, что избыточная экспрессия IFNAR приводит к взаимодействиям IFNAR1 / 2. Тем не менее, кортикальные актиновые клеточные сети и / или липидные рафты могут обеспечивать подходящий механизм для «концентрации» IFNAR для быстрой индукции устойчивых противовирусных генов всеми IFN, при этом позволяя настраивать активность, которая зависит от сродства IFN-IFNAR (124).В целом, данные предполагают, что основным механизмом, регулирующим активацию IFN, является IFN-опосредованная гетеродимеризация IFNAR1 / 2, хотя некоторые недавние данные предполагают, что вызванные IFN конформационные изменения IFNAR также могут регулировать активность IFN (125).
Семейство мышиных IFN типа I отличается от IFN человека типа I
Структура бинарного комплекса мышиных IFNβ / IFNAR1 является важным источником данных в предлагаемой модели передачи сигналов IFN типа I человека. Однако моя лаборатория и другие ранее отмечали «уникальность» семейств IFN типа I у разных животных (10, 126–129).Например, система IFN мыши состоит из 14 IFNα (обратите внимание, что обозначения подтипа IFNα мыши и человека не имеют отношения к их межвидовой последовательности и / или функциональному сходству), а также IFNβ, IFNϵ, IFNκ, лимитин (130), но не кодировать IFNω (126). Таким образом, необходимо задаться вопросом, можно ли экстраполировать мышиные IFN и рецепторные белки, а также их биологические результаты на человека. С точки зрения структурной биологии, общие складки мышиного (62) и человеческого (5) IFNβ, которые имеют 47% идентичности последовательностей, почти идентичны (рис. 6А).Внеклеточные области человеческого и мышиного IFNAR1 имеют 49% идентичности аминокислотной последовательности, и структуры доменов D1-D3 мышиного и человеческого IFNAR1 также почти идентичны (119). Эти данные позволяют предположить, что общая модель, предложенная для отсутствующего домена D4 в комплексе IFN / IFNAR2 / IFNAR1 человека, является правдоподобной (рис. 5).
Рисунок 6 Структурное сравнение IFNβ человека и мыши. (A) Структурная суперпозиция IFNβ человека (окрашена, как на рисунке 1, pdbid = 1AU1) и IFNβ мыши (пшеница, pdbid = 1WU3), подчеркивая их различные структуры петли AB. (B) Структурное наложение IFNβ мыши и человека на IFNα2 из кристаллической структуры IFNα2 / IFNAR2 человека. Полученная структурная модель приводит к стерическим конфликтам между петлей мышиного IFNβ AB и петлями связывания IFNAR2, но не для модели IFNβ / IFNAR2 человека. Этот структурный анализ обеспечивает объяснение низкой аффинности взаимодействия IFNβ / IFNAR2 мыши по сравнению с взаимодействием IFNβ / IFNAR2 человека с высокой аффинностью.
Несмотря на сходные общие структуры рецепторного комплекса, рецепторные свойства IFNβ мыши и человека различны.Человеческий IFNβ связывается с IFNAR1 и IFNAR2 со значениями ~ 30 нМ и ~ 0,1 нМ K D соответственно (28). Однако у мышей сродство к рецептору IFNβ «перевернуто», так что IFNβ-IFNAR1 образует взаимодействие с высоким сродством ( K D ~ 10 нМ), а IFNβ-IFNAR2 формирует взаимодействие с низким сродством ( K D ~ 1,7 мкМ ) (86). Структурные сравнения IFNβ человека и мыши выявляют петлю AB мышиного IFNβ, которая формирует основную часть сайта связывания IFNAR2 site-1, демонстрирует отличную структуру по сравнению с IFNβ человека (фиг. 6).В человеческом IFNβ AB-петля изгибается к N-концу спирали-F, «над» самой спиралью F, где петля соединяется со спиралью F дисульфидной связью. Напротив, AB-петля мышиного IFNβ оборачивается «поперек» спирали F, где она нарушает высокоаффинные взаимодействия IFNAR2, как это наблюдается в кристаллической структуре IFNα / IFNAR2 человека (фиг. 6B). Интересно, что выравнивание последовательностей показывает, что петли связывания мышиного рецептора IFNAR2, которые контактируют с областью петли AB мышиного IFNβ, имеют ту же длину, что и IFNAR2 человека.Кроме того, мышиные IFNα связываются с высоким сродством ( K D ~ 1 нМ) с мышиным IFNAR2 (86). Таким образом, вероятно, петли связывания мышиного рецептора IFNAR2 не изменяют своей длины или существенно не изменяют свою конформацию, чтобы приспособиться к особой структуре петли мышиного IFNβ AB. Вместе эти структурные наблюдения обеспечивают объяснение низкой аффинности взаимодействия мышиного IFNβ / IFNAR2 по сравнению с взаимодействием IFNβ-IFNAR2 человека. Хотя этот структурный анализ удовлетворителен в отношении мышиного и человеческого IFNβ, он подчеркивает многие различные свойства мышиного IFN, от структуры к механизму до исходов in vivo и , которые остаются не охарактеризованными.
Двигаясь вперед
В этом обзоре основное внимание уделяется фундаментальным структурным особенностям трех семейств IFN человека, подчеркивая сходные и уникальные особенности каждого рецепторного комплекса. Конечная цель структурных исследований — определить механизмы, которые можно использовать для открытия оптимальных терапевтических средств IFN, которые используют противовирусную активность IFN для улучшения здоровья человека (131). Важность этой цели подчеркивается пандемией SARS-CoV-2, опустошающей наше общество (72, 132–134).Основываясь на критической роли, которую сродство IFN-рецептора IFN играет в различной активности IFN (26, 120, 135), были разработаны IFN типа I и типа III с повышенным сродством к рецептору, но они не получили широкого распространения в клинике (79 , 136, 137). Предположительно потому, что мы до сих пор не знаем оптимальных принципов разработки для создания оптимального терапевтического ИФН. Учитывая, что люди продуцируют 20 различных IFN типа I / III в ответ на патогены, конструкция может быть непростой и может потребовать синергетического действия IFN типа I и типа III.Например, IFNβ типа I и IFNλ3 типа III индуцировали различные профили экспрессии антивирусных генов с различной кинетикой на гепатоцитах человека (138). В частности, высокоаффинный IFNβ индуцировал мощную противовирусную защиту почти сразу (~ 2 часа) после добавления к клеткам, которые исчезали через ~ 48 часов. Напротив, противовирусная активность IFNλ3 не наблюдалась до ~ 12 часов после лечения, но сохранялась в течение как минимум 72 часов после лечения (138). Эти данные подчеркивают взаимодействие различных рецепторных сродств и механизмов отрицательной обратной связи (139, 140), которые синергетически контролируют опосредованную IFN передачу противовирусных сигналов.Примечательно, что передача сигналов IFN типа III оказалась устойчивой к опосредованной USP18 регуляции отрицательной обратной связи, которая эффективно регулирует передачу сигналов IFN типа I (141). USP18 индуцируется IFN типа I и типа III, но специфически связывается с ICD IFNAR2 и нарушает IFNα-опосредованное образование комплекса IFNAR1 / IFNAR2. Эти исследования демонстрируют, что антивирусный сигнальный каскад, индуцированный интерферонами типа I и типа III, очень похож, однако несколько механизмов могут адаптировать ответ для достижения оптимальных функциональных результатов, включая устранение вируса и защиту хозяина.Эти и многие другие подобные исследования предоставляют новые принципы разработки для дальнейшего развития наших поисков безопасных и эффективных интерферонов с широким спектром противовирусной активности.
Вклад авторов
MRW провела литературный поиск, сделала рисунки и написала рукопись.
Финансирование
Эта рукопись частично финансировалась грантом NIH R01 AI143554.
Конфликт интересов
Автор заявляет, что исследование проводилось в отсутствие каких-либо коммерческих или финансовых отношений, которые могут быть истолкованы как потенциальный конфликт интересов.
Ссылки
2. Уилок Э. Ф. Интерфероноподобный вирус-ингибитор, индуцируемый в лейкоцитах человека фитогемагглютинином. Science (Нью-Йорк, штат Нью-Йорк) (1965) 149 (3681): 310–1. doi: 10.1126 / science.149.3681.310
CrossRef Полный текст | Google Scholar
4. Радхакришнан Р., Уолтер Л.Дж., Хруза А., Райхерт П., Тротта П.П., Нагабхушан Т.Л. и др. Опосредованный цинком димер человеческого интерферона-альфа 2b выявлен с помощью рентгеновской кристаллографии. Структура (1996) 4 (12): 1453–63.DOI: 10.1016 / S0969-2126 (96) 00152-9
PubMed Аннотация | CrossRef Полный текст | Google Scholar
5. Карпусас М., Нольте М., Бентон С.Б., Мейер В., Липскомб В.Н., Гельц С. Кристаллическая структура бета-интерферона человека при разрешении 2,2-A. Proc Natl Acad Sci U S. A (1997) 94 (22): 11813–8. doi: 10.1073 / pnas.94.22.11813
PubMed Аннотация | CrossRef Полный текст | Google Scholar
6. Иалик С.Е., Кук В.Дж., Виджай-Кумар С., Карсон М., Нагабхушан Т.Л., Тротта П.П. и др.Трехмерная структура рекомбинантного гамма-интерферона человека. Science (Нью-Йорк, штат Нью-Йорк) (1991) 252 (5006): 698–702. doi: 10.1126 / science.11
CrossRef Полный текст | Google Scholar
7. Ландар А., Карри Б., Паркер М. Х., ДиДжакомо Р., Инделикато С. Р., Нагабхушан Т. Л. и др. Дизайн, характеристика и структура биологически активного одноцепочечного мутанта IFN-гамма человека. J Mol Biol (2000) 299 (1): 169–79. doi: 10.1006 / jmbi.2000.3734
PubMed Аннотация | CrossRef Полный текст | Google Scholar
8.Рэндал М, Косяков АА. Структура и активность мономерного комплекса передачи сигналов рецептора гамма-интерферона: альфа-цепь. Struct (Camb) (2001) 9 (2): 155–63. doi: 10.1016 / S0969-2126 (01) 00567-6
CrossRef Полный текст | Google Scholar
9. Диас МО, Помикала Х.М., Боландер С.К., Малтепе Э., Малик К., Браунштейн Б. и др. Структура кластера генов интерферона I типа человека, определенная из контига клона YAC. Геномика (1994) 22 (3): 540–52. DOI: 10.1006 / geno.1994.1427
PubMed Аннотация | CrossRef Полный текст | Google Scholar
10. Пестка С. Виды интерферона-альфа человека и гибридные белки. Semin Oncol (1997) 24 (3 Suppl 9): S9–4-S9-17.
Google Scholar
12. Pestka S, Krause CD, Sarkar D, Walter MR, Shi Y, Fisher PB. Интерлейкин-10 и родственные цитокины и рецепторы. Annu Rev Immunol (2004) 22: 929–79. doi: 10.1146 / annurev.immunol.22.012703.104622
PubMed Аннотация | CrossRef Полный текст | Google Scholar
14.Hardy MP, Owczarek CM, Jermiin LS, Ejdeback M, Hertzog PJ. Характеристика локуса интерферона типа I и идентификация новых генов. Геномика (2004) 84 (2): 331–45. doi: 10.1016 / j.ygeno.2004.03.003
PubMed Аннотация | CrossRef Полный текст | Google Scholar
15. Лафлер Д.В., Нарделли Б., Царева Т., Мазер Д., Фенг П., Семенюк М. и др. Интерферон-каппа, новый интерферон типа I, экспрессируемый в кератиноцитах человека. J Biol Chem (2001) 276 (43): 39765–71.doi: 10.1074 / jbc.M102502200
PubMed Аннотация | CrossRef Полный текст | Google Scholar
17. Шеппард П., Киндсфогель В., Сюй В., Хендерсон К., Шлюцмайер С., Уитмор Т. Е. и др. IL-28, IL-29 и их рецептор цитокинов класса II IL-28R. Nat Immunol (2003) 4 (1): 63–8. doi: 10.1038 / ni873
PubMed Аннотация | CrossRef Полный текст | Google Scholar
18. Котенко С.В., Галлахер Г., Баурин В.В., Льюис-Антес А., Шен М., Шах Н.К. и др. IFN-лямбды опосредуют противовирусную защиту через особый рецепторный комплекс цитокинов класса II. Nat Immunol (2003) 4 (1): 69–77. doi: 10.1038 / ni875
PubMed Аннотация | CrossRef Полный текст | Google Scholar
19. Gad HH, Dellgren C, Hamming OJ, Vends S, Paludan SR, Hartmann R. Интерферон-лямбда функционально является интерфероном, но структурно связан с семейством интерлейкинов-10. J Biol Chem (2009) 284 (31): 20869–75. doi: 10.1074 / jbc.M109.002923
PubMed Аннотация | CrossRef Полный текст | Google Scholar
21. Walter MR. Структурный анализ членов семейства интерферонов IL-10 и I типа и их комплексов с рецептором. Adv Protein Chem (2004) 68: 171–223. doi: 10.1016 / S0065-3233 (04) 68006-5
PubMed Аннотация | CrossRef Полный текст | Google Scholar
22. Оуян В., Рутц С., Креллин Н.К., Вальдес П.А., Химовиц С.Г. Регуляция и функции цитокинов семейства IL-10 при воспалении и болезнях. Annu Rev Immunol (2011) 29: 71–109. doi: 10.1146 / annurev -munol-031210-101312
PubMed Реферат | CrossRef Полный текст | Google Scholar
24. Нагем Р.А., Колау Д., Дюмутье Л., Рено Дж. К., Огата С., Поликарпов И.Кристаллическая структура рекомбинантного интерлейкина-22 человека. Struct (Camb) (2002) 10 (8): 1051–62. doi: 10.1016 / S0969-2126 (02) 00797-9
CrossRef Полный текст | Google Scholar
25. Прокунина-Олссон Л., Мучмор Б., Тан В., Пфайфер Р. М., Парк Х., Диккеншитс Х. и др. Вариант выше IFNL3 (IL28B), создающий новый ген интерферона IFNL4, связан с нарушением клиренса вируса гепатита С. Нат Генет (2013) 45 (2): 164–71. DOI: 10.1038 / ng.2521
PubMed Аннотация | CrossRef Полный текст | Google Scholar
26.Lavoie TB, Kalie E, Crisafulli-Cabatu S, Abramovich R, DiGioia G, Moolchan K, et al. Связывание и активность всех подтипов человеческого альфа-интерферона. Цитокин (2011) 56 (2): 282–9. doi: 10.1016 / j.cyto.2011.07.019
PubMed Аннотация | CrossRef Полный текст | Google Scholar
27. Deshpande A, Putcha BD, Kuruganti S, Walter MR. Кинетический анализ сборки рецепторов, опосредованной цитокинами, с использованием сконструированных гетеродимеров FC. Protein Sci (2013) 22 (8): 1100–8. DOI: 10.1002 / про.2285
PubMed Аннотация | CrossRef Полный текст | Google Scholar
28. Jaks E, Gavutis M, Uze G, Martal J, Piehler J. Дифференциальная аффинность рецепторных субъединиц интерферонов типа I регулирует активацию дифференциального сигнала. J Mol Biol (2007) 366 (2): 525–39. doi: 10.1016 / j.jmb.2006.11.053
PubMed Аннотация | CrossRef Полный текст | Google Scholar
29. Пилер Дж., Шрайбер Г. Биофизический анализ взаимодействия человеческого ifnar2, экспрессированного в E. coli, с IFNalpha2. J Mol Biol (1999) 289 (1): 57–67. doi: 10.1006 / jmbi.1999.2726
PubMed Аннотация | CrossRef Полный текст | Google Scholar
30. Roisman LC, Jaitin DA, Baker DP, Schreiber G. Мутационный анализ сайта связывания IFNAR1 на IFNalpha2 показывает архитектуру слабого сайта связывания лиганд-рецептор. J Mol Biol (2005) 353 (2): 271–81. doi: 10.1016 / j.jmb.2005.08.042
PubMed Аннотация | CrossRef Полный текст | Google Scholar
31. Доннелли Р.П., Шейх Ф., Диккеншитс Х., Саван Р., Янг Х.А., Уолтер М.Р.Интерлейкин-26: цитокин, связанный с ИЛ-10, продуцируемый клетками Th27. Cytokine Growth Factor Rev (2010) 21 (5): 393-401. doi: 10.1016 / j.cytogfr.2010.09.001
PubMed Реферат | CrossRef Полный текст | Google Scholar
32. Доннелли Р.П., Шейх Ф., Котенко С.В., Диккеншитс Х. Расширенное семейство цитокинов класса II, которые разделяют цепь рецептора-2 IL-10 (IL-10R2). J Leukoc Biol (2004) 76 (2): 314–21. doi: 10.1189 / jlb.0204117
PubMed Аннотация | CrossRef Полный текст | Google Scholar
34.Бриско Дж., Гушин Д., Роджерс NC, Уотлинг Д., Мюллер М., Хорн Ф. и др. JAK, STAT и передача сигнала в ответ на интерфероны и другие цитокины. Philos Trans R Soc Lond B Biol Sci (1996) 351 (1336): 167–71. doi: 10.1146 / annurev.immunol.22.012703.104622
PubMed Аннотация | CrossRef Полный текст | Google Scholar
35. Ferrao R, Lupardus PJ. Домены Janus Kinase (JAK) FERM и Sh3: специфичность взаимодействий с рецептором JAK. Фронт-эндокринол (Лозанна) (2017) 8:71.doi: 10.3389 / fendo.2017.00071
PubMed Аннотация | CrossRef Полный текст | Google Scholar
37. О’Ши Дж. Дж., Шварц Д. М., Вилларино А. В., Гадина М., Макиннес И. Б., Лоуренс А. Путь JAK-STAT: влияние на болезни человека и терапевтическое вмешательство. Annu Rev Med (2015) 66: 311–28. doi: 10.1146 / annurev-med-051113-024537
PubMed Аннотация | CrossRef Полный текст | Google Scholar
40. Борден Е.С., Сен Г.К., Узе Дж., Сильверман Р.Х., Рансохофф Р.М., Фостер Г.Р. и др.Интерфероны в возрасте 50 лет: прошлое, настоящее и будущее влияние на биомедицину. Nat Rev Drug Discov (2007) 6 (12): 975–90. doi: 10.1038 / nrd2422
PubMed Аннотация | CrossRef Полный текст | Google Scholar
43. Грэм М.Б., Далтон Д.К., Гилтинан Д., Брасьале В.Л., Стюарт Т.А., Брасьале Т.Дж.. Ответ на инфекцию гриппа у мышей с направленным нарушением гена гамма-интерферона. J Exp Med (1993) 178 (5): 1725–32. doi: 10.1084 / jem.178.5.1725
PubMed Аннотация | CrossRef Полный текст | Google Scholar
44.Хуанг С., Хендрикс В., Алтаге А., Хемми С., Блюзманн Н., Камиджо Р. и др. Иммунный ответ у мышей, у которых отсутствует рецептор гамма-интерферона. Science (Нью-Йорк, штат Нью-Йорк) (1993) 259 (5102): 1742–5. doi: 10.1126 / science.8456301
CrossRef Полный текст | Google Scholar
47. Севера М., Фицджеральд К.А. TLR-опосредованная активация IFN типа I во время противовирусных иммунных ответов: борьба за победу в войне. Curr Topics Microbiol Immunol (2007) 316: 167–92. doi: 10.1007 / 978-3-540-71329-6_9
CrossRef Полный текст | Google Scholar
48.Парк А., Ивасаки А. Интерфероны типа I и типа III — индукция, сигнализация, уклонение и применение для борьбы с COVID-19. Клеточный микроб-хозяин (2020) 27 (6): 870–8. doi: 10.1016 / j.chom.2020.05.008
PubMed Аннотация | CrossRef Полный текст | Google Scholar
50. Sologuren I, Boisson-Dupuis S, Pestano J, Vincent QB, Fernandez-Perez L, Chapgier A, et al. Частичный рецессивный дефицит IFN-gammaR1: генетические, иммунологические и клинические особенности 14 пациентов из 11 родов. Hum Mol Genet (2011) 20 (8): 1509–23. DOI: 10.1093 / hmg / ddr029
PubMed Аннотация | CrossRef Полный текст | Google Scholar
51. Эрнандес Н., Буччиол Дж., Моэнс Л., Ле Пен Дж., Шахруэй М., Гудурис Э. и др. Унаследованный дефицит IFNAR1 у здоровых пациентов с побочной реакцией на живые вакцины против кори и желтой лихорадки. J Exp Med (2019) 216 (9): 2057–70. doi: 10.1084 / jem.20182295
PubMed Аннотация | CrossRef Полный текст | Google Scholar
52.Pöyhönen L, Bustamante J, Casanova J-L, Jouanguy E., Zhang Q. Опасные для жизни инфекции из-за живых ослабленных вакцин: ранние проявления врожденных ошибок иммунитета. J Clin Immunol (2019) 39 (4): 376–90. doi: 10.1007 / s10875-019-00642-3
PubMed Аннотация | CrossRef Полный текст | Google Scholar
53. Diegelmann J, Beigel F, Zitzmann K, Kaul A, Goke B, Auernhammer CJ, et al. Сравнительный анализ лямбда-интерферонов IL-28A и IL-29 в отношении их транскриптома и их противовирусных свойств против вируса гепатита С. PLoS One (2010) 5 (12): e15200. doi: 10.1371 / journal.pone.0015200
PubMed Аннотация | CrossRef Полный текст | Google Scholar
54. Zhou Z, Hamming OJ, Ank N, Paludan SR, Nielsen AL, Hartmann R. Интерферон III типа (IFN) индуцирует IFN-подобный ответ I типа в ограниченном подмножестве клеток через сигнальные пути, включающие как Путь Jak-STAT и митоген-активируемые протеинкиназы. J Virol (2007) 81 (14): 7749–58. doi: 10.1128 / JVI.02438-06
PubMed Аннотация | CrossRef Полный текст | Google Scholar
55.Смикенс С.П., Нг А., Кумар В., Джонсон М.Д., Плантинга Т.С., ван Димен С. и др. Функциональная геномика определяет путь интерферона I типа как центральный для защиты хозяина от Candida albicans. Нац Коммуна (2013) 4: 1342. doi: 10.1038 / ncomms2343
PubMed Аннотация | CrossRef Полный текст | Google Scholar
56. Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, et al. Комменсальные бактерии калибруют порог активации врожденного противовирусного иммунитета. Иммунитет (2012) 37 (1): 158–70.doi: 10.1016 / j.immuni.2012.04.011
PubMed Аннотация | CrossRef Полный текст | Google Scholar
57. Ли С., Руфаэль Н., Дурайзингем С., Ромеро-Штайнер С., Преснелл С., Дэвис С. и др. Молекулярные сигнатуры антител, полученные в результате исследования системной биологии пяти человеческих вакцин. Nat Immunol (2014) 15 (2): 195–204. doi: 10.1038 / ni.2789
PubMed Аннотация | CrossRef Полный текст | Google Scholar
58. Wu JQ, Dwyer DE, Dyer WB, Yang YH, Wang B, Saksena NK.Профили транскрипции CD8 + Т-клеток от прогрессоров ВИЧ + на ВААРТ характеризуются скоординированной активацией ферментов окислительного фосфорилирования и интерфероновыми ответами. Вирусология (2008) 380 (1): 124–35. doi: 10.1016 / j.virol.2008.06.039
PubMed Аннотация | CrossRef Полный текст | Google Scholar
59. Клаус В., Гселл Б., Лабхардт А.М., Випф Б., Сенн Х. Трехмерная структура человеческого интерферона альфа-2а с высоким разрешением, определенная методом гетероядерной ЯМР-спектроскопии в растворе. J Mol Biol (1997) 274 (4): 661–75. doi: 10.1006 / jmbi.1997.1396
PubMed Аннотация | CrossRef Полный текст | Google Scholar
60. Радхакришнан Р., Уолтер Л.Дж., Субраманиам П.С., Джонсон Х.М., Уолтер М.Р. Кристаллическая структура овечьего интерферона-тау при разрешении 2,1 А. J Mol Biol (1999) 286 (1): 151–62. doi: 10.1006 / jmbi.1998.2480
PubMed Аннотация | CrossRef Полный текст | Google Scholar
61. Вальтер MR. Трехмерные модели подтипов интерферона-альфа IFN-con1, IFN-alpha8 и IFN-alpha1, полученные из кристаллической структуры IFN-alpha2b. Semin Oncol (1997) 24 (3 Suppl 9): S9–52-S9-62.
PubMed Аннотация | Google Scholar
62. Сенда Т., Сайто С., Мицуи Ю. Уточненная кристаллическая структура рекомбинантного мышиного интерферона-бета при разрешении 2,15 А. J Mol Biol (1995) 253 (1): 187–207. doi: 10.1006 / jmbi.1995.0544
PubMed Аннотация | CrossRef Полный текст | Google Scholar
63. Ouyang S, Gong B, Li JZ, Zhao LX, Wu W, Zhang FS, et al. Структурные сведения о человеческих антителах к IFN, обладающих терапевтическим потенциалом при системной красной волчанке. J Mol Med (Berl) (2012) 90 (7): 837–46. doi: 10.1007 / s00109-012-0866-3
PubMed Аннотация | CrossRef Полный текст | Google Scholar
66. Trivella DB, Ferreira-Junior JR, Dumoutier L, Renauld JC, Polikarpov I. Структура и функция интерлейкина-22 и других членов семейства интерлейкинов-10. Cell Mol Life Sci (2010) 67 (17): 2909–35. doi: 10.1007 / s00018-010-0380-0
PubMed Аннотация | CrossRef Полный текст | Google Scholar
67. Boniface K, Guignouard E, Pedretti N, Garcia M, Delwail A, Bernard FX и др.Роль интерлейкина 22, полученного из Т-клеток, в псориатическом воспалении кожи. Clin Exp Immunol (2007) 150 (3): 407–15. doi: 10.1111 / j.1365-2249.2007.03511.x
PubMed Аннотация | CrossRef Полный текст | Google Scholar
68. Brand S, Beigel F, Olszak T, Zitzmann K, Eichhorst ST, Otte JM, et al. IL-22 увеличивается при активной болезни Крона и способствует экспрессии провоспалительных генов и миграции кишечных эпителиальных клеток. Am J Physiol Gastrointest Liver Physiol (2006) 290 (4): G827–38.doi: 10.1152 / ajpgi.00513.2005
PubMed Аннотация | CrossRef Полный текст | Google Scholar
70. Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, et al. Интерлейкин-22 обеспечивает раннюю защиту хозяина от прикрепления и уничтожения бактериальных патогенов. Nat Med (2008) 14 (3): 282–9. DOI: 10,1038 / нм1720
PubMed Аннотация | CrossRef Полный текст | Google Scholar
72. Prokunina-Olsson L, Alphonse N, Dickenson RE, Durbin JE, Glenn JS, Hartmann R, et al.COVID-19 и возникающие вирусные инфекции: случай лямбда-интерферона. J Exp Med (2020) 217 (5): 1–4. doi: 10.1084 / jem.20200653
CrossRef Полный текст | Google Scholar
73. Major J, Crotta S, Llorian M, McCabe TM, Gad HH, Priestnall SL, et al. Интерфероны типа I и III нарушают восстановление эпителия легких во время выздоровления от вирусной инфекции. Sci (N Y NY) (2020) 369 (6504): 712–7. doi: 10.1126 / science.abc2061
CrossRef Полный текст | Google Scholar
74.Broggi A, Ghosh S, Sposito B, Spreafico R, Balzarini F, Lo Cascio A и др. Интерфероны III типа разрушают эпителиальный барьер легких после распознавания вирусом. Science (Нью-Йорк, штат Нью-Йорк) (2020) 369 (6504): 706–12. doi: 10.1126 / science.abc3545
CrossRef Полный текст | Google Scholar
75. Grajales-Reyes GE, Colonna M. Реакции интерферона при вирусных пневмониях. Sci (N Y NY) (2020) 369 (6504): 626–7. doi: 10.1126 / science.abd2208
CrossRef Полный текст | Google Scholar
77.Зданов А., Шалк-Хихи С., Густчина А., Цанг М., Уэтерби Дж., Влодавер А. Кристаллическая структура интерлейкина-10 обнаруживает функциональный димер с неожиданным топологическим сходством с гамма-интерфероном. Структура (1995) 3 (6): 591–601. DOI: 10.1016 / S0969-2126 (01) 00193-9
PubMed Аннотация | CrossRef Полный текст | Google Scholar
78. Котенко С.В. Семейство цитокинов, связанных с ИЛ-10, и их рецепторы: родственны, но в какой степени? Cytokine Growth Factor Rev (2002) 13 (3): 223-40.DOI: 10.1016 / S1359-6101 (02) 00012-6
PubMed Аннотация | CrossRef Полный текст | Google Scholar
79. Mendoza JL, Schneider WM, Hoffmann HH, Vercauteren K, Jude KM, Xiong A, et al. Комплекс IFN-λ-IFN-λR1-IL-10Rβ обнаруживает структурные особенности, лежащие в основе функциональной пластичности IFN типа III. Иммунитет (2017) 46 (3): 379–92. doi: 10.1016 / j.immuni.2017.02.017
PubMed Аннотация | CrossRef Полный текст | Google Scholar
81. Sommereyns C, Paul S, Staeheli P, Michiels T.IFN-lambda (IFN-lambda) экспрессируется тканезависимым образом и в первую очередь действует на эпителиальные клетки in vivo . PLoS Pathog (2008) 4 (3): e1000017. doi: 10.1371 / journal.ppat.1000017
PubMed Аннотация | CrossRef Полный текст | Google Scholar
82. Mordstein M, Neugebauer E, Ditt V, Jessen B, Rieger T, Falcone V, et al. Лямбда-интерферон делает эпителиальные клетки дыхательных путей и желудочно-кишечного тракта устойчивыми к вирусным инфекциям. Дж. Вирол (2010) 84 (11): 5670–7.doi: 10.1128 / jvi.00272-10
PubMed Аннотация | CrossRef Полный текст | Google Scholar
84. Pott J, Mahlakoiv T, Mordstein M, Duerr CU, Michiels T, Stockinger S, et al. IFN-лямбда определяет противовирусную защиту кишечного эпителия хозяина. Proc Natl Acad Sci U S. A (2011) 108 (19): 7944–9. DOI: 10.1073 / pnas.1100552108
PubMed Аннотация | CrossRef Полный текст | Google Scholar
85. Fung KY, Mangan NE, Cumming H, Horvat JC, Mayall JR, Stifter SA и др.Интерферон-эпсилон защищает женские половые пути от вирусных и бактериальных инфекций. Science (Нью-Йорк, штат Нью-Йорк) (2013) 339 (6123): 1088–92. doi: 10.1126 / science.1233321
CrossRef Полный текст | Google Scholar
86. Стифтер С.А., Мэтьюз А.Ю., Манган Н.Э., Фунг К.Ю., Дрю А., Тейт М.Д. и др. Определение отличительных внутренних свойств нового интерферона I типа, IFN. J Biol Chem (2018) 293 (9): 3168–79. doi: 10.1074 / jbc.M117.800755
PubMed Аннотация | CrossRef Полный текст | Google Scholar
87.Куре Дж., Таскер С., Ким Дж., Сихвонен Т., Фрутвала С., Куэйл А. Дж. И др. Дифференциальная регуляция экспрессии генов IFNalpha, IFNbeta и IFNepsilon в эпителиальных клетках шейки матки человека. Cell Biosci (2017) 7:57. doi: 10.1186 / s13578-017-0185-z
PubMed Аннотация | CrossRef Полный текст | Google Scholar
88. Нарделли Б., Зарицкая Л., Семенюк М., Чо Ю. Х., Лафлер Д. В., Шах Д. и др. Регулирующий эффект IFN-каппа, нового IFN типа I, на продукцию цитокинов клетками врожденной иммунной системы. J Immunol (Балтимор, Мэриленд: 1950) (2002) 169 (9): 4822–30. doi: 10.4049 / jimmunol.169.9.4822
CrossRef Полный текст | Google Scholar
89. Харрис Б.Д., Шрейтер Дж., Шевриер М., Джордан Дж. Л., Уолтер М.Р. Человеческий интерферон-ε и интерферон-каппа проявляют низкую активность и низкое сродство к IFNAR на клеточной поверхности и антагонисту поксвируса B18R. J Biol Chem (2018) 293 (41): 16057–68. doi: 10.1074 / jbc.RA118.003617
PubMed Аннотация | CrossRef Полный текст | Google Scholar
90.Микнис З.Дж., Маграчева Э., Ли В., Зданов А., Котенко С.В., Влодавер А. Кристаллическая структура человеческого интерферона-лямбда1 в комплексе с его высокоаффинным рецептором интерферон-лямбдаR1. J Mol Biol (2010) 404 (4): 650–64. doi: 10.1016 / j.jmb.2010.09.068
PubMed Аннотация | CrossRef Полный текст | Google Scholar
91. Юн С.И., Джонс BC, Логсдон, штат Нью-Джерси, Харрис Б.Д., Дешпанде А., Радаева С. и др. Структура и механизм разделения рецепторов общей цепью IL-10R2. Структура (2010) 18 (5): 638–48.doi: 10.1016 / j.str.2010.02.009
PubMed Аннотация | CrossRef Полный текст | Google Scholar
92. Деллгрен К., Гад Х. Х., Хэмминг О. Дж., Мельхьорсен Дж., Хартманн Р. Интерферон-лямбда 3 человека является мощным членом семейства интерферонов III типа. Genes Immun (2009) 10 (2): 125–31. doi: 10.1038 / gene.2008.87
PubMed Аннотация | CrossRef Полный текст | Google Scholar
93. Terczyńska-Dyla E, Bibert S, Duong FHT, Krol I, Jørgensen S, Collinet E, et al. Снижение активности IFNλ4 связано с улучшенным клиренсом HCV и снижением экспрессии генов, стимулированных интерфероном. Нац Коммуна (2014) 5 (1): 5699. doi: 10.1038 / ncomms6699
PubMed Аннотация | CrossRef Полный текст | Google Scholar
94. Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, et al. Генетическая изменчивость IL28B позволяет прогнозировать клиренс вируса, вызванный лечением гепатита С. Nature (2009) 461 (7262): 399–401. DOI: 10.1038 / nature08309
PubMed Аннотация | CrossRef Полный текст | Google Scholar
95. Танака Ю., Нисида Н., Сугияма М., Куросаки М., Мацуура К., Сакамото Н. и др.Полногеномная ассоциация IL28B с ответом на терапию пегилированным интерфероном-альфа и рибавирином хронического гепатита C. Nat Genet (2009) 41 (10): 1105–9. DOI: 10.1038 / ng.449
PubMed Аннотация | CrossRef Полный текст | Google Scholar
96. Suppiah V, Moldovan M, Ahlenstiel G, Berg T., Weltman M, Abate ML, et al. IL28B связан с ответом на терапию интерфероном-альфа и рибавирином при хроническом гепатите С. Нат Генет (2009) 41 (10): 1100–4. DOI: 10,1038 / нг.447
PubMed Аннотация | CrossRef Полный текст | Google Scholar
97. Хэмминг OJ, Terczyńska-Dyla E, Vieyres G, Dijkman R, Jørgensen SE, Akhtar H, et al. Интерферон лямбда 4 сигнализирует через рецептору IFNλ для регулирования противовирусной активности против ВГС и коронавирусов. EMBO J (2013) 32 (23): 3055–65. doi: 10.1038 / emboj.2013.232
PubMed Аннотация | CrossRef Полный текст | Google Scholar
98. Мегер А., Висвалингам К., Дилгер П., Брайан Д., Вадхва М.Биологическая активность интерлейкинов-28 и -29: сравнение с интерферонами I типа. Цитокин (2005) 31 (2): 109–18. doi: 10.1016 / j.cyto.2005.04.003
PubMed Реферат | CrossRef Полный текст | Google Scholar
99. Mendoza JL, Escalante NK, Jude KM, Sotolongo Bellon J, Su L, Horton TM, et al. Структура рецепторного комплекса IFNγ определяет дизайн предвзятых агонистов. Nature (2019) 567 (7746): 56–60. doi: 10.1038 / s41586-019-0988-7
PubMed Аннотация | CrossRef Полный текст | Google Scholar
100.Thomas C, Moraga I, Levin D, Krutzik PO, Podoplelova Y, Trejo A, et al. Структурная связь между дискриминацией лиганда и активацией рецептора интерферонами типа I. Ячейка (2011) 146 (4): 621–32. doi: 10.1016 / j.cell.2011.06.048
PubMed Аннотация | CrossRef Полный текст | Google Scholar
101. Вальтер М.Р., Виндзор В.Т., Нагабхушан Т.Л., Ланделл Д.Д., Ланн Калифорния, Заудни П.Дж. и др. Кристаллическая структура комплекса между гамма-интерфероном и его растворимым высокоаффинным рецептором. Nature (1995) 376 (6537): 230–5. doi: 10.1038 / 376230a0
PubMed Аннотация | CrossRef Полный текст | Google Scholar
102. Walter MR. Молекулярная основа функции IL-10: от рецепторной структуры до начала передачи сигналов. Текущие темы Microbiol Immunol (2014) 380: 191–212. doi: 10.1007 / 978-3-662-43492-5_9
CrossRef Полный текст | Google Scholar
103. Mao X, Ren Z, Parker GN, Sondermann H, Pastorello MA, Wang W и др. Структурные основы ассоциации нефосфорилированного STAT1 и рецепторного связывания. Mol Cell (2005) 17 (6): 761–71. doi: 10.1016 / j.molcel.2005.02.021
PubMed Аннотация | CrossRef Полный текст | Google Scholar
104. Чен Х, Винкемайер У., Чжао Й, Джерузалми Д., Дарнелл Дж. Мл., Куриан Дж. Кристаллическая структура фосфорилированного тирозином димера STAT-1, связанного с ДНК. Cell (1998) 93 (5): 827–39. doi: 10.1016 / s0092-8674 (00) 81443-9
PubMed Аннотация | CrossRef Полный текст | Google Scholar
105. Krause CD, Lunn CA, Izotova LS, Mirochnitchenko O, Kotenko SV, Lundell DJ, et al.Сигнализация ковалентными гетеродимерами гамма-интерферона. Доказательства односторонней передачи сигналов в активном тетрамерном рецепторном комплексе. J Biol Chem (2000) 275 (30): 22995–3004. doi: 10.1074 / jbc.M7199
PubMed Аннотация | CrossRef Полный текст | Google Scholar
107. О’Доннелл Л.А., Хенкинс К.М., Кулкарни А., Матулло С.М., Балачандран С., Паттисапу А.К. и др. Гамма-интерферон индуцирует защитные неканонические сигнальные пути в первичных нейронах. J Neurochem (2015) 135 (2): 309–22.doi: 10.1111 / jnc.13250
PubMed Аннотация | CrossRef Полный текст | Google Scholar
108. Ивашкив Л.Б. IFNγ: передача сигналов, эпигенетика и роль в иммунитете, метаболизме, иммунотерапии болезней и рака. Nat Rev Immunol (2018) 18 (9): 545–58. doi: 10.1038 / s41577-018-0029-z
PubMed Аннотация | CrossRef Полный текст | Google Scholar
109. Натан К.Ф., Мюррей Х.В., Вибе М.Э., Рубин BY. Идентификация гамма-интерферона как лимфокина, который активирует окислительный метаболизм макрофагов человека и антимикробную активность. J Exp Med (1983) 158 (3): 670–89. doi: 10.1084 / jem.158.3.670
PubMed Аннотация | CrossRef Полный текст | Google Scholar
110. Янг Х.А., Лещ JH. IFN-гамма: последние достижения в понимании регуляции экспрессии, биологических функций и клинического применения. Curr Topics Microbiol Immunol (2007) 316: 97–117. doi: 10.1007 / 978-3-540-71329-6_6
CrossRef Полный текст | Google Scholar
112. Альспах Э., Люсье Д.М., Шрайбер Р.Д. Интерферон γ и его важная роль в стимулировании и подавлении спонтанного и терапевтического иммунитета к раку. Cold Spring Harbor Perspect Biol (2019) 11 (3): 1–20. doi: 10.1101 / cshperspect.a028480
CrossRef Полный текст | Google Scholar
113. Zhang SY, Boisson-Dupuis S, Chapgier A, Yang K, Bustamante J, Puel A, et al. Врожденные ошибки опосредованного интерфероном (IFN) иммунитета у людей: понимание соответствующих ролей IFN-альфа / бета, IFN-гамма и IFN-лямбда в защите хозяина. Immunol Rev (2008) 226: 29–40. doi: 10.1111 / j.1600-065X.2008.00698.x
PubMed Аннотация | CrossRef Полный текст | Google Scholar
114.Chill JH, Quadt SR, Levy R, Schreiber G, Anglister J. Рецептор интерферона человека типа I: структура ЯМР раскрывает молекулярную основу связывания лиганда. Struct (Camb) (2003) 11 (7): 791–802. doi: 10.1016 / S0969-2126 (03) 00120-5
CrossRef Полный текст | Google Scholar
115. Нудельман И., Акабеков С.Р., Шерф Т., Англистер Дж. Наблюдение межмолекулярных взаимодействий в больших белковых комплексах с помощью двумерной двойной разностной ядерной спектроскопии Оверхаузера: приложение к комплексу интерферон-рецептор 44 кДа. J Am Chem Soc (2011) 133 (37): 14755–64. doi: 10.1021 / ja205480v
PubMed Аннотация | CrossRef Полный текст | Google Scholar
116. Нудельман И., Акабеков С.Р., Шнур Э., Бирон З., Леви Р., Сюй Й и др. Межмолекулярные взаимодействия в комплексе 44 кДа интерферон-рецептор, обнаруженные с помощью асимметричного обратного протонирования и двумерного NOESY. Биохимия (2010) 49 (25): 5117–33. doi: 10.1021 / bi100041f
PubMed Аннотация | CrossRef Полный текст | Google Scholar
117.Пилер Дж., Шрайбер Г. Мутационный и структурный анализ интерфейса связывания между интерферонами типа I и их рецептором Ifnar2. J Mol Biol (1999) 294 (1): 223–37. doi: 10.1006 / jmbi.1999.3230
PubMed Аннотация | CrossRef Полный текст | Google Scholar
118. Li Z, Strunk JJ, Lamken P, Piehler J, Walz T. ЭМ-структура комплекса интерферон-рецептор типа I раскрывает новый механизм передачи сигналов цитокинов. J Mol Biol (2008) 377 (3): 715–24. DOI: 10.1016 / j.jmb.2007.12.005
PubMed Аннотация | CrossRef Полный текст | Google Scholar
119. Де Верд Н. А., Вивиан Дж. П., Нгуен Т. К., Манган Н. Э., Гулд Дж. А., Бранифф С. Дж. И др. Структурная основа уникальной оси передачи сигналов интерферона-бета опосредована через рецептор IFNAR1. Nat Immunol (2013) 14 (9): 901–7. doi: 10.1038 / ni.2667
PubMed Аннотация | CrossRef Полный текст | Google Scholar
120. Kalie E, Jaitin DA, Podoplelova Y, Piehler J, Schreiber G.Стабильность тройного комплекса интерферон-рецептор, а не сродство к отдельным субъединицам диктует различную биологическую активность. J Biol Chem (2008) 283 (47): 32925–36. doi: 10.1074 / jbc.M806019200
PubMed Аннотация | CrossRef Полный текст | Google Scholar
121. Levin D, Schneider WM, Hoffmann HH, Yarden G, Busetto AG, Manor O, et al. Многогранная активность интерферона I типа выявляется антагонистом рецепторов. Научный сигнал (2014) 7 (327): ra50.doi: 10.1126 / scisignal.2004998
PubMed Аннотация | CrossRef Полный текст | Google Scholar
122. Krause CD, Digioia G, Izotova LS, Xie J, Kim Y, Schwartz BJ, et al. Лиганд-независимое взаимодействие рецепторного комплекса интерферона I типа необходимо для наблюдения за его биологической активностью. Cytokine (2013) 64 (1): 286–97. doi: 10.1016 / j.cyto.2013.06.309
PubMed Аннотация | CrossRef Полный текст | Google Scholar
123. Wilmes S, Beutel O, Li Z, Francois-Newton V, Richter CP, Janning D, et al.Динамика димеризации рецепторов как регулятор пластичности передачи сигналов интерферона I типа. J Cell Biol (2015) 209 (4): 579–93. doi: 10.1083 / jcb.201412049
PubMed Аннотация | CrossRef Полный текст | Google Scholar
124. You C, Marquez-Lago TT, Richter CP, Wilmes S, Moraga I, Garcia KC, et al. Стабилизация димера рецептора с помощью иерархических микрокомпартментов плазматической мембраны регулирует передачу сигналов цитокинов. Sci Adv (2016) 2 (12): e1600452. DOI: 10.1126 / sciadv.1600452
PubMed Аннотация | CrossRef Полный текст | Google Scholar
125. Sharma N, Longjam G, Schreiber G. Передача сигналов интерферона I типа не связана с ориентацией конкретного рецептора посредством мягких требований трансмембранного домена. J Biol Chem (2016) 291 (7): 3371–84. doi: 10.1074 / jbc.M115.686071
PubMed Аннотация | CrossRef Полный текст | Google Scholar
126. van Pesch V, Lanaya H, Renauld JC, Michiels T. Характеристика семейства генов альфа-интерферона мыши. J Virol (2004) 78 (15): 8219–28. doi: 10.1128 / jvi.78.15.8219-8228.2004
PubMed Аннотация | CrossRef Полный текст | Google Scholar
129. van Pesch V, Michiels T. Характеристика интерферона-альфа 13, нового конститутивного подтипа мышиного интерферона-альфа. J Biol Chem (2003) 278 (47): 46321-8. doi: 10.1074 / jbc.M302554200
PubMed Аннотация | CrossRef Полный текст | Google Scholar
130. Оритани К., Медина К.Л., Томияма Ю., Исикава Дж., Окадзима Ю., Огава М. и др.Лимитин: интерфероноподобный цитокин, который преимущественно влияет на предшественников В-лимфоцитов. Nat Med (2000) 6 (6): 659–66. doi: 10.1038 / 76233
PubMed Аннотация | CrossRef Полный текст | Google Scholar
133. Felsenstein S, Herbert JA, McNamara PS, Hedrich CM. COVID-19: иммунология и варианты лечения. Clin Immunol (Орландо, Флорида) (2020) 215: 108448. doi: 10.1016 / j.clim.2020.108448
CrossRef Полный текст | Google Scholar
134. Wu Z, McGoogan JM.Характеристики и важные уроки вспышки коронавирусного заболевания 2019 г. (COVID-19) в Китае: сводка отчета о 72314 случаях, полученного Китайским центром по контролю и профилактике заболеваний. Джама (2020) 323 (13): 1239–42. doi: 10.1001 / jama.2020.2648
PubMed Аннотация | CrossRef Полный текст | Google Scholar
135. Jaitin DA, Roisman LC, Jaks E, Gavutis M, Piehler J, Van der Heyden J, et al. Изучение дифференциального действия интерферонов (IFN): мутант IFN-альфа2 с повышенным сродством к IFNAR1 функционально подобен IFN-бета. Mol Cell Biol (2006) 26 (5): 1888–97. doi: 10.1128 / MCB.26.5.1888-1897.2006
PubMed Аннотация | CrossRef Полный текст | Google Scholar
136. Калие Э., Джайтин Д.А., Абрамович Р., Шрайбер Г. Мутант интерферона альфа2, оптимизированный с помощью фагового дисплея для связывания IFNAR1, обеспечивает специфически усиленную противоопухолевую активность. J Biol Chem (2007) 282 (15): 11602–11. doi: 10.1074 / jbc.M610115200
PubMed Аннотация | CrossRef Полный текст | Google Scholar
137.Brideau-Andersen AD, Huang X, Sun SC, Chen TT, Stark D, Sas IJ, et al. Направленная эволюция генно-перетасованных молекул IFN-альфа с профилями активности, адаптированными для лечения хронических вирусных заболеваний. Proc Natl Acad Sci U S. A (2007) 104 (20): 8269–74. DOI: 10.1073 / pnas.060
04PubMed Аннотация | CrossRef Полный текст | Google Scholar
138. Forero A, Ozarkar S, Li H, Lee CH, Hemann EA, Nadjsombati MS, et al. Дифференциальная активация фактора транскрипции IRF1 лежит в основе различных иммунных ответов, вызываемых интерферонами типа I и типа III. Иммунитет (2019) 51 (3): 451–64.e6. doi: 10.1016 / j.immuni.2019.07.007
PubMed Аннотация | CrossRef Полный текст | Google Scholar
139. Porritt RA, Hertzog PJ. Динамическое управление сигнализацией IFN типа I с помощью интегрированной сети отрицательных регуляторов. Trends Immunol (2015) 36 (3): 150–60. doi: 10.1016 / j.it.2015.02.002
PubMed Аннотация | CrossRef Полный текст | Google Scholar
141. Francois-Newton V, Magno de Freitas Almeida G, Payelle-Brogard B, Monneron D, Pichard-Garcia L, Piehler J, et al.Контроль отрицательной обратной связи на основе USP18 индуцируется интерферонами типа I и типа III и специфически инактивирует ответ интерферона альфа. PLoS One (2011) 6 (7): e22200. doi: 10.1371 / journal.pone.0022200
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Потенциальная роль интерферонов в лечении пациентов с COVID-19
https://doi.org/10.1016/j.intimp.2020.107171Получить права и контентОсновные моменты
- •
Кризисы COVID-19 возникают по распространение тяжелого острого респираторного синдрома коронавируса 2 (SARS-CoV-2).
- •
SARS-CoV-2 может вызывать от слабых до тяжелых респираторных заболеваний и даже смерть пациентов.
- •
Разработка эффективной терапии является неотложной задачей для борьбы с пандемией SARS-CoV-2.
- •
IFN оказались решающими в борьбе с SARS-CoV-2 у пациентов с COVID-19.
- •
Комбинированная терапия может быть более эффективной для лечения этого заболевания, чем монотерапия.
Реферат
Недавний кризис общественного здравоохранения в мире вызван распространением коронавируса 2 (SARS-CoV-2), также известного как COVID-19.Вирус происходит от летучих мышей и переносится к людям через неопределенных промежуточных животных. Этот вирус может вызывать от слабых до тяжелых респираторных заболеваний, включая острый респираторный дистресс-синдром (ARDS), синдром полиорганной недостаточности (MODS), пневмонию и даже смерть пациентов. Болезнь COVID-19 распространяется при вдыхании через зараженные капли или при контакте с зараженной средой. Время инкубации составляет от 2 до 14 дней, и симптомы обычно включают лихорадку, боль в горле, кашель, недомогание, утомляемость, одышку и другие.Следует учитывать, что многие инфицированные люди протекают бессимптомно. Несколько лабораторий поддерживают разработку различных иммунологических и вирусологических методов диагностики этого заболевания. Лечение в основном поддерживающее; однако есть несколько агентов, которые используются для лечения пациентов с COVID-19. Интерфероны (IFN) имеют решающее значение в борьбе с COVID-19 и могут быть подходящим кандидатом для лечения этих пациентов. Комбинированная терапия может быть более эффективной для лечения этого заболевания, чем монотерапия.Профилактика требует изолирования подозреваемых людей и домашнего карантина, а также оказания помощи инфицированным людям с легкой или тяжелой формой заболевания в больницах. Поскольку вспышка SARS-CoV-2 ускорилась, разработка эффективной терапии является неотложным требованием для борьбы с вирусом и предотвращения дальнейшей пандемии. В этой рукописи мы рассмотрели доступную информацию о SARS-CoV-2 и возможных методах лечения пациентов с COVID-19.
Ключевые слова
SARS-CoV-2
COVID-19
Респираторный коронавирус
Пневмония
Интерферон
Рекомендуемые статьи Цитирующие статьи (0)
© 2020 Elsevier B.V. Все права защищены.
Рекомендуемые статьи
Цитирующие статьи
Ротавирусная инфекция индуцирует как интерферон (IFN), так и резистентность к воспалению путем перепрограммирования передачи сигналов кишечного рецептора IFN
Введение
Ротавирусы представляют собой сегментированные вирусы дцРНК без оболочки, которые вызывают тяжелую дегидратирующую диарею у многих млекопитающих. , в результате чего ежегодно умирает около 200 000 младенцев (1). Острая инфекция RV у гомологичного хозяина (естественный вид, у которого штамм RV реплицируется с высоким титром и распространяется от человека к человеку) является чрезвычайно эффективным процессом; тяжелая диарея может быть вызвана воздействием очень маленького инфекционного инокулята (2).Ответ на врожденный интерферон хозяина (IFN) представляет собой многостороннюю линию защиты от вирусных патогенов, поскольку его индукция может активировать сотни различных антивирусных генов и запускать воспалительные сигналы, тем самым устраняя инфицированные клетки, ограничивая распространение вирусной инфекции на неинфицированные клетки-свидетели, и формирование как величины, так и качества последующего адаптивного иммунного ответа (3–5). Индукция всех трех основных типов IFN (α / β, γ и λ ) происходит во время гомологичной инфекции RV тонкой кишки (6–10), но противовирусные эффекты IFN-ответа на гомологичную репликацию RV в лучшем случае скромны (10).Несмотря на то, что IFN-опосредованные воспалительные реакции плохо изучены, они также, вероятно, уменьшаются при инфекции RV, которая связана только с легким воспалением кишечника (11, 12) по сравнению с другими острыми диарейными патогенами (13). Как RV эффективно подрывает способность различных типов IFN выполнять свои противовирусные и воспалительные функции (14), не совсем понятно.
С другой стороны, инфицирование гетерологичными RV (RV, как правило, не выделяется из инфицированных видов хозяев) обычно приводит к сильно ограниченному репликации в кишечнике и незначительной или отсутствующей устойчивой передаче неинфицированным хозяевам (11, 15, 16).У мышей-сосунков репликация некоторых гетерологичных штаммов RV в кишечнике существенно ограничена (~ 10 4 раз), но может быть частично устранена удалением рецепторов IFN или нижележащего конвергентного фактора транскрипции STAT1, тем самым демаскируя ключевую роль IFN в подавлении Инфекционность RV у гетерологичных видов хозяев (7, 10, 11, 16). Истощение IFNR у грудных мышей также может привести к острому воспалительному заболеванию желчевыводящих путей (часто летальному) после инфицирования гетерологичным штаммом Rhesus RV (RRV) (11).Таким образом, определение механизмов, с помощью которых гомологичные RV подавляют противовирусные и воспалительные реакции хозяина IFN в кишечнике, имеет жизненно важное значение для разработки более эффективных стратегий борьбы с заболеваниями и смертностью, связанными с RV, и для понимания патогенного потенциала естественно циркулирующих штаммов RV.
Результаты
Гомологичная инфекция RV ограничивает ответы амплификации IFN в кишечнике
Мы сравнили репликацию вируса в кишечнике и противовирусные ответы между ограниченным репликацией, IFN-чувствительным штаммом обезьян (RRV) и компетентным к репликации, IFN-устойчивым RV мыши. штамм (EW) (10) при 12 и 72 hpi с помощью qRT-PCR (рис. 1A-B).Оба штамма RV сравнимо индуцировали три основных транскрипта IFN-типа (IFN-α / β, IFN-γ и IFN- λ , фиг. 1A) при 12 hpi. Сходная индукция путей IFN обоими штаммами RV поддерживается на уровне белка путем измерения кишечного IFN- γ , который продуцируется как у EW, так и у RRV RV-инфицированных детенышей мыши при 24 hpi (фиг. 1C). Некоторые IFN-стимулированные гены (ISG) также транскрипционно индуцировались на 12 hpi с аналогичной эффективностью как EW, так и RRV ( IRF7 , ISG15 , IFIh2 , DDX58 , IRF9 , IFIT2 909 MX2 ).
Рисунок 1. Влияние репликационно-компетентных (гомологичных, мышиных) и репликационно-ограниченных (гетерологичных, обезьяньих) штаммов RV на кишечный интерферон-стимулированный ген (ISG) и воспалительные реакции транскрипции.(A-B) Транскрипционный анализ кишечных транскрипционных ответов на штаммы EW мышей и RRV RV обезьян. Сосущих мышей в возрасте от трех до пяти дней (N = 4 на группу) инфицировали мышиными штаммами EW или обезьяньими штаммами RRV RV (или ложными), и через 12 часов или 72 часов тонкий кишечник собирали для транскрипционного анализа хозяина (A). и гены RV (B).(C) Кишечную секрецию IFN-γ измеряли с помощью Luminex после инфицирования RV через 24 часа на дюйм, как в (A) (N = 3 образца на группу, каждый образец объединен от 3 детенышей; ND, не обнаружено).
Несмотря на индукцию IFNs и различных транскриптов ISG с аналогичной или более низкой эффективностью, уровни транскрипта NSP5 EW RV в кишечнике были заметно выше, чем RRV при 12 hpi (более чем в 10 4 раз, рис. 1B). Удивительно, но через 72 hpi индукция нескольких типов IFN и транскриптов ISG оставалась сопоставимой у мышей, инфицированных мышиным или обезьяньим RV (рис.1A и фиг. S1). Однако репликация EW RV на 72 hpi оставалась существенно выше, чем RRV RV (в ~ 10 4 раз, фиг. 1B и фиг. S2). Единственным исключением из этих результатов был IL28A (кодирующий IFN- λ 2), который был значительно выше (~ 10 раз) у детенышей, инфицированных EW RV, при 72 hpi. Что касается кинетики, индукция IFNs типа I ( IFNA4 и IFNA5 ) и IRF7 транскриптов обоими штаммами RV значительно увеличивалась от 12 до 72 hpi, в то время как индукция IFN типа II ( IFNG ) и типа III ( IL28A ) снизился в течение периода инфицирования.Следует отметить, что 12-часовая индукция нескольких генов, стимулированных интерфероном, и воспалительные реакции транскрипции у детенышей, инфицированных EW RV, были практически устранены через 72 hpi ( IL6 , CXCL10 , ISG20 , IFI203 , PELI1 , IFIh2 , DDX58 , IRF9 , IFIT2 , 72 hpi, фиг. 1A), напоминающие ответы амплификации IFN на некомпетентные к репликации RRV RV. Таким образом, кишечные ответы амплификации IFN на EW и RRV не отражают заметных различий в фактической репликации вируса между этими двумя типами RV.Вероятное объяснение этих результатов состоит в том, что, несмотря на существенную продолжающуюся репликацию EW RV в кишечнике и индукцию генов IFN в течение первых 3 дней инфекции, гомологичный мышиный RV подавлял амплификацию кишечных ISG и воспалительных генов на 72 hpi.
Репликация гомологичного ротавируса подавляет расщепление нескольких каспаз в кишечнике.
Приведенные выше результаты показали, что некоторые IFN продолжают индуцироваться транскрипционно устойчиво в течение 72 часов во время гомологичной инфекции RV.Воздействие на клетки IFN (IFN-α / β и —γ ) в течение длительного времени обычно приводит к последовательному расщеплению инициаторной каспазы 8 и исполнительной каспазы 3, способствуя апоптозу (17–20). Как гомологичный RV борется с потенциалом опосредованной каспазой гибели клеток при устойчивой индукции различных кишечных типов IFN (рис. 1A, 1C), изучено недостаточно. Поэтому мы затем исследовали экспрессию предшественников (полноразмерных, FL) и расщепленных (Cl) форм инициатора каспазы 8, которая имеет решающее значение для внешней гибели клеток (21), у мышей, инфицированных EW RV.Иммуноблоттинг IEC тонкой кишки показал, что инфекция правого желудочка приводит к существенно пониженному соотношению изоформ Cl: FL каспазы 8 (2,2 в имитации по сравнению с 0,5 при инфицировании правого желудочка через 24 часа на дюйм, но не при 3 часах на дюйм) (рис. 2А). Предполагается, что дефектное расщепление каспазы 8 (фиг. 2A) нарушит последующее расщепление апоптотической каспазы 3-исполнителя, поэтому мы затем исследовали статус каспазы 3 во время инфицирования RV. Как и ожидалось, кишечник мышей-сосунков, инфицированных RV, также показал пониженное расщепление каспазы 3 (Cl.: Коэффициенты FL 1,4 и 0,4 у ложных и EW RV инфицированных, соответственно, при 24 hpi, но не 3 hpi) (рис. 2B). Эти неожиданные результаты показывают, что гомологичная инфекция RV ингибирует кишечные каспазы на стадии их активации зимогена.
Рисунок 2. Ротавирус подавляет расщепление нескольких каспаз и способствует расщеплению взаимодействующей с рецептором киназы протеина 1 (RIP1K).(A-B) Анализ экспрессии каспазы 8 и каспазы 3. Детенышей мышей перорально инокулировали EW (R) или имитировали (M).Через 3 и 24 часа на дюйм основные кишечные клетки собирали из цельных тканей тонкого кишечника и обрабатывали для SDS-PAGE. Гели анализировали вестерн-блоттингом на предшественники (FL) и расщепленные (Cl.) Изоформы либо каспазы 8 (A), либо каспазы 3 (B), а также других белков. Нормализованные отношения Cl. к каспазам FL, определенные денситометрией, указаны ниже на отдельных панелях. (C) FACS-анализ расщепления каспазы 8 и каспазы 3. Резюме анализа проточной цитометрии CD45 — CD44 — ESA + IEC после внутриклеточного окрашивания на KI67 и расщепленной каспазы 8 или расщепленной каспазы 3 от инфицированных EW RV или контрольных детенышей при 24 hpi.(D) Процент IEC (CD45 — CD44 — ESA + ), демонстрирующих расщепленную каспазу 8 или расщепленную каспазу 3 у ложных и инфицированных EW мышей при 24 hpi, усредненный по результатам трех экспериментов; Планки погрешностей представляют собой стандартные ошибки средних значений. (E) Внутриклеточное окрашивание на структурный белок RV (VP6) и расщепленную каспазу 8 (верхняя панель) или расщепленная каспаза 3 (нижняя панель) в CD45 — CD44 — ESA + IEC после EW RV или имитационной инфекции в 24 hpi.(F) Иммуноблоттинг-анализ ткани тонкого кишечника от детенышей мышей, инфицированных EW, RRV или имитатором, и собранных в указанные моменты времени. (G) Анализ расщепления каспазой 9 в культивируемых фибробластах NIH 3T3, инфицированных IFN-стимулирующим штаммом RV, Великобритания. Клетки 3T3 инфицировали бычьим UK RV (или имитацией) в течение указанного времени, а затем анализировали с помощью иммуноблоттинга. Цифры под пятнами указывают нормализованные отношения Cl. к каспазе 9 FL. (H) IEC HT-29 человека инфицировали SB1-A RV свиньи (или имитатором) в течение указанного времени и анализировали с помощью иммуноблоттинга.Цифры под пятнами указывают нормализованные отношения Cl. к каспазе 8 FL. (I) Клетки HT-29, инфицированные указанными штаммами RV, анализировали на RIP1K, RIP3K и VP6 при 4 или 12 hpi с помощью иммуноблоттинга. (J) Клетки HT-29 были инфицированы SB1-A RV (или имитацией) или обработаны TNF-альфа (10 нг / мл) и циклогексимидом (10 мкг / мл) в течение 4 часов, а затем проанализированы на клеточную адгезию или фосфатидилсерин. маркерное выражение. Показанные данные являются репрезентативными для двух или более независимых экспериментов.
Расщепление каспаз 8 и 3 ингибируется как в инфицированных ротавирусом, так и в неинфицированных случайных ворсинчатых энтероцитах
Наблюдая, что инфекция RV in vivo приводит к нарушению процессинга каспазы-8 и -3 в IEC, мы затем исследовали, является ли этот процесс возникает, в частности, в эпителии ворсинок тонкой кишки (7).Используя проточную цитометрию, мы регистрировали живые ворсинчатые IEC, используя ранее описанную комбинацию маркеров клеточной поверхности (например, CD45 — ESA + CD44 — ) (7, 22), а затем измеряли экспрессию расщепленных форм любой из каспаз. 8 или каспаза 3 в RV инфицированных и ложных контрольных детенышах (рис. 2C). Эти эксперименты подтвердили существенное снижение расщепления каспазы 8 и каспазы 3 в ворсинчатых энтероцитах тонкого кишечника во время инфекции RV (~ 75% снижение расщепления каспазы 8 и каспазы 3 после инфекции).
Затем мы исследовали, происходит ли ингибирование расщепления каспаз только в инфицированных RV зрелых ворсинчатых IEC через 24 часа на дюйм, используя панель антител для определения типа клеток, активности каспазы и положительности VP6 RV (в качестве маркера статуса клеток в отношении инфицирования RV). (Рис. 2D). Примечательно, что эффективное подавление расщепления каспазой-8 происходит как в популяции зрелых ворсинчатых клеток VP6 + (инфицированные), так и в VP6 — (т.е. неинфицированные «свидетели»). Относительные соотношения клеток, положительных по расщепленной каспазе 8, в различных популяциях составляли: ~ 3.0 (имитация), 0,2 (прохожие) и 0,6 (инфицированный RV) (рис. 2D, верхние панели). Эффективный ингибирующий эффект инфекции RV на неинфицированные случайные клетки (т.е. VP6 —) также наблюдался после анализа расщепления каспазы 3 с использованием аналогичного подхода (рис. 2D, относительные отношения расщепления каспазой 3 составляли 4,0, 0,3 и 0,9 в псевдониме). , VP6 — случайных и VP6 + RV-инфицированных клеток, соответственно; рис. 2D, нижние панели).
Ожидается, что ингибирование RV расщепления каспаз 8 и 3 как в инфицированных RV, так и в неинфицированных IEC тонкой кишки окажет значительное влияние на жизнеспособность кишечных клеток.Поэтому мы исследовали гибель клеток в двух клеточных компартментах кишечника. Во-первых, анализ жизнеспособности суспензий единичных клеток тонкого кишечника (т.е. «основной» уровень) выявил умеренное общее снижение гибели кишечных клеток во время инфекции EW RV (это снижение не было статистически значимым) (рис. S3A-B). Во-вторых, при окрашивании TUNEL клеток кишечного негематопоэтического (т.S3C, p <0,001). В совокупности эти результаты демонстрируют, что инфекция RV in vivo эффективно предотвращает расщепление каспаз и подавляет гибель кишечных клеток как в инфицированных, так и в неинфицированных клетках-свидетелях.В отличие от внешнего апоптоза, опосредованного каспазой 8, инициатор каспаза 9 расщепляется во время внутреннего апоптоза (23). Интересно, что мыши-сосунки, инфицированные EW RV, также демонстрировали нарушение расщепления каспазы 9 (рис. 2E), на что указывает накопление кишечной FL каспазы 9 на 16hpi, но не на 6hpi, по сравнению с RRV или ложно-инфицированными контролями (рис. .2E). Чтобы изучить роль IFN в этом процессе, мы определили, блокируется ли расщепление каспазы 9 в культивируемых фибробластах 3T3, инфицированных штаммом RV крупного рогатого скота UK, который, как мы ранее показали, запускает MAVS-зависимую секрецию IFN-β в этой клеточной линии (24, 25). Заражение клеток 3T3 UK RV в течение 9 часов подавляло расщепление каспазы 9 и опосредовало накопление зимогена FL-каспазы 9. Напротив, повышенная экспрессия Cl-каспазы 9 и снижение экспрессии FL-каспазы 9 происходили при 16 hpi (фиг. 2F). Эти результаты подтвердили, что ингибирующие эффекты RV на внутреннюю активность инициатора каспазы 9 не требуют ингибирования секреции IFN, и дополнительно показали, что расщепление каспазы может регулироваться во время инфекции RV.
Кинетическая регуляция каспаз с помощью RV была дополнительно исследована на линии клеток кишечного происхождения путем анализа динамики расщепления каспазы 8 в культивируемых клетках HT-29 после инфицирования штаммом свиного RV SB1-A, который, как мы ранее показали, предотвращает IFN- секреция β в этой клеточной линии (26). По сравнению с имитационно инфицированным контролем, инфицирование SB1-A RV ингибировало расщепление каспазы 8, начиная с 4 hpi, но с максимальным эффектом при 8 hpi (фиг. 2G). В совокупности эти исследования показывают, что RV в целом подавляет события инициаторного расщепления каспазы, существенные как для внешней, так и для внутренней гибели клеток, и регулирует эти процессы во времени во время инфекции.Результаты также продемонстрировали, что регуляторные эффекты RV на передачу сигналов апоптоза как in vitro, (фиг. 2F-G), так и in vivo (фиг. 2A-E) проявляются независимо от его способности подавлять индукцию IFN.
Несколько штаммов RV индуцируют расщепление регулятора клеточной гибели RIP1K
В ответ на длительное воздействие IFN расщепление каспазы 8 приводит к апоптотическому отщеплению эпителиальных клеток с обычно легкими воспалительными последствиями (20). Напротив, в клетках, дефектных по расщеплению каспазой 8 (например, IEC во время инфицирования EW RV in vivo , рис.2A-C), стимуляция IFN обычно направляет быструю и стремительную форму гибели клеток в результате некроптоза, который является острым воспалительным процессом (17, 27). Рецептор-взаимодействующая протеин-1 киназа (RIP1K) является важной адапторной киназой с двойной функцией на перекрестке решения клетки жить или умереть в ответ на апоптотические или некроптотические стимулы (28, 29). Эти противоположные функции RIP1K в основном регулируются протеолитическим механизмом (30, 31). В то время как сверхэкспрессия полноразмерного человеческого RIP1K запускает как апоптоз, так и некроптоз, опосредованное каспазой расщепление RIP1K ~ 75 кДа (p75) на нескольких сайтах в пределах киназного домена (KD) и домена взаимодействия (ID) генерирует более короткие фрагменты, полученные либо из RIP1K N-концевой (51 кДа, 37 кДа и 25 кДа) или С-концевой (39 кДа) области.Как p51, так и p39 RIP1K эффективно ингибируют функцию p75 RIP1K и расщепление каспазы 8, предотвращая апоптотическую и некроптотическую гибель клеток в ответ на устойчивые воспалительные стимулы (31).
Поскольку RV регулирует расщепление каспазы 8 во времени во время инфекции (Fig. 2F-G), мы исследовали, подвергается ли расщепление RIP1K также регуляции RV. Клетки HT-29 были инфицированы репрезентативными штаммами RV, различающимися по способам регуляции IFN in vitro (RRV обезьяны, деградирующей IRF3, SB1-A свиньи, ингибирующей NF-kB, и человеческий WI-61, и мутантный по NSP1 обезьяний SA11-5S. Штамм RV, который стимулирует секрецию IFN-β (32)) в течение 4 или 12 часов, а затем анализируется иммуноблоттингом с использованием анти- (N-концевого) антитела RIP1K (рис.2H). По сравнению с ложно инфицированными клетками HT-29 инфицирование различными штаммами RV приводило к усилению экспрессии p75 RIP1K на 4 hpi с различной эффективностью, и эта повышенная экспрессия также выявлялась позже во время инфицирования (на 12 hpi). Кроме того, инфекция RV также генерировала несколько более коротких фрагментов расщепления RIP1K p51 / p37 / p25, и в уровнях этих фрагментов расщепления были очевидны штамм-специфические различия. Примечательно, что все штаммы RV, включая SA11-5S, запускали расщепление p51-RIP1K, начинающееся на ранней стадии инфицирования, за исключением инфицирования штаммом RV человека WI-61, где повышенная экспрессия p51 RIP1K в первую очередь обнаруживается с помощью 12hpi.Более того, штаммы обезьян (RRV и SA11-5S) и человека (WI-61) RV индуцировали дополнительное расщепление RIP1K (p37 / p25 RIP1K — RRV и SA11-5S; p37 RIP1K — WI-61). Интересно, что SB1-A RV индуцирует повышенную экспрессию как (про-смерть) полноразмерной изоформы p75 RIP1K, так и более короткой (ингибирующей) изоформы p51 RIP1K (фиг. 2H, дорожки 5-6). Таким образом, мы исследовали, как функционально антагонистические фрагменты RIP1K (p75 и p51), которые экспрессируются во время инфекции SB1-A, влияют на гибель клеток. Мы обнаружили, что как адгезия клеток, так и экспрессия фосфатидилсерина (маркер апоптозных клеток) при 16 hpi в SB1-A RV-инфицированных клетках напоминают ложно инфицированные HT-29 клетки, предполагая, что повышенная экспрессия p75 RIP1K не вызывает гибель клеток.Напротив, клетки HT-29, обработанные смесью фактора некроза опухоли и циклогексимида, демонстрировали значительную потерю клеточной адгезии и повышенное апоптотическое окрашивание (фиг. 2I). Эти результаты показывают, что накопление RIP1K и его протеолиз регулируются во время инфекции RIP1K, и более короткая доминантно-отрицательная регуляторная изоформа RIP1K p51 генерируется многими RV на ранних стадиях инфекции, что потенциально может нарушать апоптоз и некроптоз.
Ротавирус ограничивает кишечные IFN-опосредованные воспалительные реакции
Помимо стимулирования IFNR-опосредованной и каспазозависимой воспалительной смерти, различные типы IFN также могут управлять выработкой воспалительных цитокинов с помощью IFNR-направленного процесса амплификации гена с положительной прямой связью ( 3, 33).Поэтому мы затем исследовали, подавляет ли гомологичная RV-инфекция грудных мышей, помимо ингибирования активации каспазы in vivo , синтез продуктов гена-мишени воспаления, стимулированного IFNR. Чтобы рассмотреть эту возможность, мы использовали экспериментальную схему, аналогичную рис. 1A, для измерения уровней продукции различных кишечных цитокинов с разрешением 1 dpi у сосущих мышей, инфицированных либо гомологичными мышиными (штамм EW), либо гетерологичными обезьянами (штамм RRV) RV (рис. 3A). ). Эти исследования показывают значительное увеличение продукции различных цитокинов в кишечнике в ответ на инфекцию гомологичных EW и / или гетерологичных RRV RV (RANTES, IP-10, IL1-A, EOTAXIN, GRO-A, MCP-3, MIP-1B). , IL-4, GM-CSF, IL-15, LIF; рис.3А). Из них набор цитокинов (MCP-3, IL-15, LIF, IL-10, MCP-1, MIP-2α и IL-18) был сравнительно повышен в ответ на репликационно-компетентные мышиные (EW) или ограниченный репликацией обезьяний (RRV) RV. Напротив, второй набор преимущественно провоспалительных цитокинов (RANTES, IP-10, IL1-α, Eotaxin, GRO-α, MIP-1b, IL-6, IL-4, GM-CSF и IL-17A) в первую очередь индуцировались у гетерологичных обезьяньих RRV-инфицированных детенышей. Эти цитокины коррелируют с плохой кишечной репликацией гетерологичных RV у мышей, хотя еще предстоит определить, вносят ли они какой-либо вклад в ограничение роста RRV.Поразительно, что несколько дифференциально секретируемых цитокинов в этом наборе, как сообщается, являются эффекторами, опосредованными IFN-α / β, IFN-, γ и / или IFN- λ (т.е. RANTES (34–36), IP-10 (35, 37), эотаксин (38), GRO-α (39, 40) и MIP-1b (36, 41, 42)). Это говорит о том, что успешная репликация в кишечнике гомологичного мышиного правого желудочка сопровождается подавлением IFNR-амплифицированных воспалительных цитокиновых ответов (14).
Рисунок 3. Влияние репликационно-компетентных (гомологичных, мышиных) и репликационно-ограниченных (гетерологичных, обезьяньих) штаммов RV на продукцию цитокинов в кишечнике.(A) Секреция кишечных цитокинов во время инфицирования RV мышей-сосунков. Сосущих мышей инфицировали, как показано на Фигуре 1, и гомогенаты тонкой кишки анализировали через 24 часа на дюйм на предмет указанных уровней цитокинового белка (N = 3 образца на группу, каждый образец объединяли от 3 детенышей и измеряли в двух экземплярах). (B) Схема описанного эндотоксин-опосредованного воспаления, которое стимулируется IFNR и негативно регулируется расщепленной каспазой 8. (C) Влияние гомологичной инфекции RV у мышей-сосунков на эндотоксин-опосредованное повреждение кишечника.Мыши-сосунки были ложно инфицированы или инфицированы EW RV, и через 3 дня им парентерально вводили очищенный эндотоксин (15 мкг на детеныша) в течение 6 часов перед забором тонкого кишечника для гистологического исследования путем окрашивания гематоксилином и эозином. Представленные данные являются репрезентативными для 2 детенышей.
Чтобы подтвердить, подавляет ли гомологичный мышиный RV воспаление, усиленное IFNR, мы использовали модель эндотоксемии на мышах-сосунках, в которой LPS-индуцированное воспаление и тяжелое поражение кишечника, как сообщается, возникают в основном через IFNR-опосредованную экспрессию воспалительных цитокинов (43–1). 45) и некроптотической гибели клеток (17, 27, 46).В предыдущей работе мы обнаружили, что инфицирование RV мышей тонкой кишки защищает сосущих мышей от смертности, вызванной эндотоксинами (8). Однако не оценивалось, оказывает ли репликация EW RV местный защитный эффект в тонкой кишке мышей, зараженных LPS. Здесь мы специально исследовали, предотвращает ли инфекция EW RV воспалительное повреждение тонкого кишечника во время эндотоксемии (рис. 3B). Мышам-сосункам, инфицированным перорально EW RV (или имитатором) в течение 72 часов, систематически вводили эндотоксин, и через 6 часов исследовали возникшее повреждение кишечника.У ложно инфицированных мышей парентеральное введение эндотоксина приводило к тяжелому кишечному некрозу, травмам и разрушению тканей через 6 часов, как и ожидалось (рис. 3C) (47). Напротив, мыши-сосунки, инфицированные мышиным EW RV (для 3d) перед провокацией эндотоксином, демонстрируют минимальное повреждение тонкого кишечника (фиг. 3C). Эти результаты согласуются с выводом о том, что гомологичный RV эффективно предотвращает воспалительное повреждение тонкого кишечника, усиленное рецептором IFN, из-за некроптоза или экспрессии воспалительных генов.Результаты также показывают, несколько неожиданно, что эффективное ингибирование каспазы 8 в тонком кишечнике во время EW RV-инфекции (фиг. 2, C, D) не повышает чувствительность мышей к LPS-индуцированному воспалению.
Различные штаммы RV усиливают резистентность к IFN в неинфицированных HT-29 клетках-свидетелях
Расщепленная каспаза 8 является ключевым негативным регулятором IFN (и LPS) -опосредованного некроптоза и острого воспаления (17), а делеция каспазы 8, специфичная для IEC, приводит к спонтанное воспаление кишечника (48), возможно, вызванное конститутивной тонической передачей сигналов IFN (27, 46).Таким образом, ингибирование расщепления каспазы 8 считается мощным инициирующим событием для острого IFN-направленного некроптотического воспаления в кишечнике (17, 21, 46). Хотя инфекция RV мыши снижает расщепление изоформ каспазы 8 как в инфицированных, так и в сторонних IEC (рис. 2D) и индуцирует различные кишечные IFN in vivo (7, 9) (рис. 1A), эти последствия не приводят к заметным последствиям. Амплификация ISG, воспаление кишечника или повреждение тканей (рис. 1A, 3A-C), гибель клеток (рис. S3) или ограничение роста вируса (рис.1Б) (7, 10, 11). Чтобы лучше понять этот очевидный парадокс, мы исследовали, может ли инфекция правого желудочка блокировать передачу сигналов рецептора IFN (IFNR) как в инфицированных, так и в клетках-свидетелях, чтобы эффективно предотвращать эффекторные функции IFN. Ранее мы сообщали, что в культуре клеток несколько штаммов RV опосредуют эффективную лизосомную деградацию эндогенных субъединиц IFNAR1, IFNGR1 и IFNLR1 в отдельных инфицированных RV + клетках, но не в неинфицированных клетках-свидетелях RV (8).
Мы предположили, что RV может использовать другую стратегию для отключения функций IFNR в незараженных сторонних клетках.В предыдущих исследованиях in vitro мы показали, что штамм свиного RV (SB1-A) (но не несколько других штаммов RV) ингибировал IFNR-опосредованное фосфорилирование STAT1-Y701 в клетках-свидетелях RV (26) после относительно короткого ( т.е. 30-минутная) стимуляция высокими концентрациями очищенного IFNβ (т.е. до 2000 МЕ / мл). Здесь мы более подробно исследовали, могут ли другие штаммы RV также подавлять реактивность IFN в клетках-свидетелях, но с меньшей эффективностью, чем штамм свиней SB1-A.В этом повторном исследовании использовалось увеличенное время введения для стимуляции экзогенного IFN перед анализом STAT1-pY701, таким образом, лучше учитывала возможное снижение эффективности ингибирования IFNR другими штаммами RV (фиг. 4A). Клетки HT-29 человека были инфицированы несколькими штаммами RV (обезьяний RRV и SA11, свиной SB1-A, бычий UK и монореассортант SOF обезьяны SA11, кодирующий свиной NSP1) в течение 12 часов, а затем клетки стимулировали в течение 6 часов очищенным IFN- β (500 МЕ / мл) перед анализом экспрессии STAT1-pY701 в отдельных клетках RV-VP6 + и VP6 — HT-29.Стимуляция ложно инфицированных клеток IFN-β активировала фосфорилирование STAT1-Y701, и эта активация была эффективно ингибирована всеми протестированными штаммами RV в инфицированных RV клетках VP6 + , как мы сообщали ранее (26). Что еще более важно, активация STAT1-Y701 также значительно ингибировалась в клетках-свидетелях RV-VP6 — другими штаммами RV (фиг. 4A). Таким образом, эти результаты показывают, что все испытанные штаммы RV ингибируют передачу сигналов рецептора IFN в популяции сторонних клеток in vitro , хотя их относительная эффективность, по-видимому, варьируется в зависимости от штамма.
Рисунок 4. Ротавирус усиливает устойчивость к опосредованной рецептором IFN активации STAT1 и транскрипционной функции.(A) Различные штаммы RV вызывают устойчивость к IFN-опосредованному фосфорилированию STAT1-Y701 в клетках-свидетелях. Клетки HT-29 инфицировали указанными штаммами RV и через 12 часов стимулировали 500 МЕ / мл очищенного IFNβ в течение 6 часов перед анализом FACS. Цифры в красном цвете указывают на кратное увеличение STAT1-pY701 после стимуляции IFN в популяциях RV VP6 + (инфицированных) и VP6- (сторонних наблюдателей).Данные представляют два независимых эксперимента. (B-C) Влияние инфекции RV на транскрипционную активность, опосредованную dsRNA- и IFN-рецепторами. На схеме вверху показан использованный экспериментальный подход. Клетки HT-29 были инфицированы RV (штамм RRV, при MOI ~ 0,5) или ложно-инфицированы, и при 6 hpi клетки либо трансфицировали 2 мкг очищенной дцРНК, либо стимулировали 500 МЕ / мл очищенного IFNβ. При 12 hpi клетки анализировали с помощью qRT-PCR на экспрессию транскриптов хозяина (B) и RV (C).Данные представляют собой два независимых эксперимента. (D) Схематическое обобщение дифференциального ингибирования транскрипции, стимулированной дцРНК и рецептором IFN, с помощью RV. Пунктирная красная линия указывает на возможную передачу сигналов отрицательной обратной связи при эктопической стимуляции IFNR во время инфицирования RV, сплошные красные линии указывают на ингибирование IFNR в RV за счет деградации рецептора (в RV-инфицированных клетках (8)) и за счет блокирования IFNR-опосредованной активации и транскрипции STAT1 (в Прохожие на фургоне).
Поскольку разные типы IFNR, обычно экспрессируемые в клетках-свидетелях правого желудочка (8), вероятно, будут функционально нарушены (рис.4A), мы затем исследовали, как RV-ингибирование IFNR-опосредованного фосфорилирования STAT1 влияет на транскрипционные функции последующих антивирусных и воспалительных генов. Некоторые ISG и воспалительные транскрипты, активируемые стимуляцией IFNR-лигандом, также могут быть индуцированы, если клетки стимулируются внутриклеточной дцРНК, в первую очередь через сигнальные пути NF-kB и IRF3 (49). Таким образом, здесь мы сравнили способность RV регулировать клеточный ISG и воспалительную транскрипционную амплификацию после стимуляции либо экзогенным IFN-β, либо внутриклеточной длинной дцРНК (рис.4Б). Контрольные клетки HT-29, стимулированные IFN-β или дцРНК, запускали как перекрывающиеся ( TNF, TNFAIP3, HERC5, IFI6, IFIT3, IFIT1, IFNB1, RSAD2 ), так и уникальные ( TLR2, IL26, IL21, SERPING посредством IFN-β. ; IL29, IFNG только дцРНК) наборов транскрипционных ответов. Независимо от того, как осуществлялась стимуляция транскрипции, инфекция RV эффективно предотвращала дальнейшую активацию транскрипции HERC5, IFNG, IL22, IL21, IFI6, IFIT1, IFIT3 и RSAD2 .Напротив, мы идентифицировали набор провоспалительных транскриптов, которые специфически блокировались RV в ответ на экзогенный IFN-β, но не на внутриклеточную dsRNA ( TLR2, IL26, TNF, IRF8, TNFAIP3, IL29, IL1B) . Среди измеренных транскриптов только SERPING эффективно индуцировался стимуляцией IFNAR1 во время инфицирования RV, хотя и с меньшей эффективностью, чем в неинфицированных клетках HT-29. Стимулирующее ингибирование транскрипции RV не было связано с чувствительностью репликации RV, поскольку оно не изменялось ни в условиях стимуляции IFN-β, ни в условиях стимуляции дцРНК (рис.4С). Кроме того, индукция определенных транскриптов специфически подавлялась эктопической стимуляцией IFNAR1 во время инфекции RV ( HERC5, IFNG, и IL22, статистически значимо, p <0,01 ; IRF8, IL29, и IL1B, не статистически значимо. ). Эти результаты демонстрируют, что RV эффективно ингибирует опосредованную IFN-β-рецептором транскрипцию выбранных ISG и воспалительных генов. Кроме того, они указывают на то, что эктопическая стимуляция IFNAR1 во время инфекции RV может вызывать ингибирование транскрипции по отрицательной обратной связи (рис.4Б, 4Д). Такая регуляция на основе обратной связи (50), вероятно, особенно актуальна для противовирусных и воспалительных функций неинфицированных RV-клеток-свидетелей, которые, в отличие от RV + инфицированных клеток, экспрессируют IFNR (8) (рис. 4D).
Инфекция RV у мышей повторно направлена на IFNR-направленную кишечную противовирусную и воспалительную передачу сигналов
Сообщается, что стимуляция IFNGR1 и IFNAR1 подавляет STAT1 посредством передачи сигналов отрицательной обратной связи при определенных условиях, способствуя устойчивости к IFN в контексте рака и хронической вирусной инфекции (33, 50).Мы исследовали, изменяется ли кишечная IFNR-опосредованная транскрипция гомологичным RV in vivo для подавления противовирусной и воспалительной передачи сигналов IFN (рис. 1A и 3A). После заражения мышей-сосунков мышиным EW RV в течение 12 часов мы парентерально вводили очищенный IFN-γ (или PBS) и собирали тонкий кишечник через дополнительные 12 часов для транскрипционного анализа (фиг. 5A). У ложно инфицированных контрольных детенышей стимуляция IFN- γ активировала несколько кишечных транскриптов ( STAT1, ISG15, IFI204, IL10RA, IL1RN, статистически значимо ; S100A8, BIRC5, REG3G, не значимо; фиг.5Б-5Д). Напротив, транскрипция CCL5 значительно подавлялась эктопической стимуляцией IFNGR1. Подобные транскрипционные ответы также активируются инфекцией EW RV, но с величиной, значительно большей, чем полученная при стимуляции только IFN-γ . Набор противовирусных ( DHX58, DDX58, MX2 ) и воспалительных ( TRAIL, NOS2, CXCL10 ) транскриптов значительно усиливался RV, но не стимуляцией IFN-γ . Примечательно, что эктопическая стимуляция IFNGR1 у мышей, инфицированных EW RV, либо значительно подавлялась ( STAT1, ISG15, TRAIL, и CXCL10 ), либо полностью устранялась ( DHX58, DDX58, MX2, CCL5, и NOS2 ). кишечные противовирусные и воспалительные реакции (рис. 5B-C).Напротив, стимулированные IFN-γ детеныши, инфицированные EW RV, активировали транскрипцию противовоспалительных и пролиферативных генов ( S100A8, BIRC5, ARG2, REG3G, MKI67, и IL1RN ) (фиг. 5D). Эти транскрипты не индуцировались в значительной степени ни стимуляцией IFN-γ , ни одной только инфекцией EW RV, и синергетически увеличивались, когда стимуляция IFNGR1 осуществлялась во время инфицирования EW RV. Чтобы выяснить, можно ли объяснить эффекты стимуляции IFN у крысят, инфицированных EW RV, измененными уровнями репликации RV, мы измерили РНК RV в кишечнике с помощью qRT-PCR.Результаты демонстрируют, что репликация EW RV существенно не изменилась после стимуляции IFN-γ (фиг. 5E). Эти результаты предполагают, что во время гомологичной RV-инфекции функция кишечного IFNR может быть направлена на подавление противовирусных и воспалительных реакций и стимулирование противовоспалительной программы, что позволяет выявить потенциальный механизм, с помощью которого неинфицированные RV-клетки-свидетели противостоят противовирусным и воспалительным эффектам IFN, секретируемого IEC. кишечные кроветворные клетки (7, 51).
Рисунок 5. Ротавирусная инфекция повторно использует управляемую рецептором IFN кишечную и воспалительную передачу сигналов.(A) Схема использованного экспериментального подхода. (B-E) Транскрипционный анализ различных категорий кишечных транскриптов после эктопической стимуляции рецептора IFN типа II. Мышей инфицировали EW RV (или имитацией) (N = 3-5 детенышей на группу) и через 12 hpi вводили очищенный мышиный IFN γ (1 мкг на детеныша или PBS, внутрибрюшинно). Двенадцать часов спустя мышей умерщвляли и РНК тонкого кишечника анализировали с помощью qRT-PCR на различные транскрипты гена NSP5 хозяина (B-D) или RV NSP5 (E).
Обсуждение
Интерфероны представляют собой плейотропные эффекторы врожденного иммунитета, которые составляют основную линию защиты от вирусных патогенов. Посредством передачи сигналов через свои родственные рецепторы различные типы IFN усиливают экспрессию многочисленных и многофункциональных противовирусных и воспалительных генов (4, 5). Кроме того, стимуляция IFNR может также вызывать апоптотические и некроптотические формы гибели клеток (17–19, 46), возможно, для ограничения репликации и распространения вируса. У гомологичных видов-хозяев репликация RV лишь незначительно ограничивается индукцией различных IFN, а надежная вирусная репликация не связана со значительным воспалением (7, 8, 10, 11, 16).Однако репликация гетерологичного RV сильно ограничена каждым из трех основных типов IFN (10). Как гомологичным RV удается избежать противовирусного и воспалительного действия IFN, до сих пор полностью не изучено.
Мы обнаружили, что у мышей-сосунков врожденный иммунный транскрипционный ответ слизистой оболочки на вирулентный и высокоинфекционный штамм EW RV мышей очень напоминал ответ на ограниченный репликацией гетерологичный штамм обезьян (рис. 1А). Сходство врожденных ответов, включая транскрипцию типа IFN, продукцию IFN-γ и амплификацию ISG или транскриптов воспалительных генов с течением времени (рис.1C) — плохо коррелировали с очень разными уровнями репликации EW и RRV (разница примерно в 10 000 раз, рис. 1B). Транскрипты различных типов IFN оставались значимо повышенными на 72 hpi у детенышей, инфицированных EW RV, что свидетельствует об устойчивом врожденном иммунном ответе на продолжающуюся репликацию RV. Однако мы не наблюдали амплификацию нескольких ISG (т.е. ISG15, ISG20, IFI203, IFIh2, DDX58, IRF9 и MX2 (рис. 1A) на более поздних стадиях заражения EW.Эти результаты показали, что, хотя гомологичная репликация RV и индукция IFN происходят устойчиво во время инфекции, это не приводит эффективно к устойчивой амплификации множества противовирусных и воспалительных генов.
В ответ на длительное воздействие IFN может происходить апоптоз за счет протеолитической активации инициатора каспазы 8 (19, 20). IFN-стимулированный апоптоз — это относительно медленный процесс, вероятно, опосредованный IFN-стимулированной экспрессией промежуточных апоптотических лигандов (например,g., FasL и TRAIL) (18–20) и обычно имеет легкие воспалительные последствия. Помимо запуска апоптоза, протеолитическое расщепление прокаспазы 8 может подавлять некроптоз — более воспалительную форму гибели клеток (17, 52). Некроптоз быстро активируется при дефиците в клетках каспазы 8 или в присутствии ингибиторов каспаз после воздействия воспалительных лигандов, таких как TNF-α или FasL (21). Стимуляция лигандом рецепторов IFN-α / β и —γ также может вызывать быстрый клеточный некроптоз, приводящий к острому воспалению (17, 21).Этот процесс опосредуется неизвестными в настоящее время, но критическими IFNR-зависимыми эффекторами (называемыми постоянно IFN-регулируемыми эффекторами некроптоза, CIREN) (46). Кроме того, IFN могут усиливать экспрессию генов воспалительных цитокинов, кодирующих IFN-чувствительные элементы промотора ответа. И IFN-опосредованный некроптоз, и экспрессия воспалительных генов требуют наличия интактного сигнального пути IFNR и являются критическими медиаторами острого кишечного воспаления в ответ на эндотоксин (17, 21, 43, 46, 53). Следует отметить, что до этой серии экспериментов регулируемая гибель клеток и воспалительные ответвления передачи сигналов рецептора IFN не были хорошо изучены в контексте инфекции RV.
Наши результаты предполагают, что гомологичная инфекция правого желудочка инициирует двойной набор событий, чтобы подорвать и задержать опосредованные IFN функции гибели клеток и потенциально предоставить больше времени для репликации правого желудочка в IEC. По оценкам, 1400 клеток на ворсинки в день обычно выделяются для поддержания гомеостаза кишечника у взрослой мыши (54). Мы обнаружили, что ключевой этап обновления эпителиальных клеток, расщепление каспаз 8 и 3, значительно ингибировался во время гомологичной инфекции RV как в инфицированных вирусом, так и в неинфицированных сторонних зрелых ворсинчатых IEC (рис.2, S3). Это блокирование активации каспаз может служить для предотвращения или уменьшения IFN-ускоренного апоптоза и, таким образом, для усиления вирусной репликации и распространения на неинфицированные IEC. Ингибирование активности каспазы 3 само по себе может быть результатом ингибируемого RV расщепления вышележащей инициаторной каспазы 8 (рис. 2A, 2C-D), которая опосредует активацию каспазы 3 внешним путем гибели и ингибируется как в RV-инфицированных, так и у сторонних наблюдателей. МЭК (рис. 2D). Кроме того, гомологичный мышиный RV также препятствует гомеостатическому расщеплению другой инициаторной каспазы (каспазы 9), которая имеет решающее значение для реализации внутренних путей клеточной смерти.Способность RV регулировать несколько типов каспаз in vivo является новой и, насколько нам известно, представляет собой оригинальную вирусную стратегию для широкой регуляции и задержки нескольких эффекторов гибели кишечных клеток. Ингибирование расщепления каспаз также происходит во время инфицирования RV in vitro и регулируется во времени (фиг. 2F, G).
Механизм, с помощью которого расщепление различных типов каспаз регулируется RV, в настоящее время неизвестен. Тем не менее, анализ ключевого эффектора передачи сигналов, регулируемого каспазой 8, RIP1K (28, 30, 53), в RV-инфицированных клетках HT-29 дает возможное понимание основных механизмов.В частности, мы обнаружили, что in vitro различных штаммов RV способствовали расщеплению полноразмерного p75 RIP1K на более короткие фрагменты (т.е. p51, p37 и p25) (рис. 2H). Расщепление RIP1K на фрагмент p51 опосредуется каспазой 8 и генерирует негативный регулятор, который может предотвращать апоптотическую и некроптотическую гибель клеток в клетках, постоянно подвергающихся действию воспалительных стимулов (31). В отличие от регуляции RIP1K каспазой 8, при определенных условиях (например, стрессе ER) RIP1K необходим для правильного расщепления каспазы 8 и апоптоза (55).Это указывает на существование регуляторной обратной связи RIP1K на активность каспазы 8. Одним из возможных объяснений наших находок может быть то, что один или несколько продуктов расщепления RIP1K, генерируемых на ранних этапах во время инфекции RV (Fig. 2H), регулируют расщепление каспазы 8 и гибель клеток во время стрессовых состояний ER, которые возникают позже при инфекции RV (56-59). Анализ динамики расщепления каспазы 8 и RIP1K во время инфицирования RV (рис. 2G-H) показал, что оба этих процесса нарушены во времени, но не опосредуют заметных уровней гибели клеток или маркеров апоптоза даже на относительно поздних сроках инфицирования (рис.2I). Будет интересно определить, происходят ли эти регуляторные процессы RIP1K в клетках-свидетелях RV, и идентифицировать вовлеченные эффекторы вируса и хозяина.
Независимо от точных лежащих в основе механизмов, нарушение расщепления каспазы 8 как в RV инфицированных, так и в сторонних IECs (Fig. 2D) создает потенциально проблемную ситуацию в кишечном эпителии (21). МЭК-специфической делеции каспазы 8 достаточно, чтобы вызвать спонтанные воспалительные поражения кишечника, приводящие к терминальному илеиту и колиту (48, 60).Как отмечалось выше, ингибирование расщепления каспазы 8 также является ключевым событием прайминга, которое запускает опосредованный рецептором IFN некроптоз, острое воспаление и повреждение тканей, вызванное эндотоксином (17-19, 27, 46, 53, 60). Парадоксально, но мы обнаружили, что инфекция EW RV у мышей значительно защищала грудных мышей от последующей летальной эндотоксемии (выживаемость 100% против 0% через 24 часа после воздействия эндотоксина) (8). Поскольку эндотоксин-опосредованное повреждение усиливается экспрессией воспалительного гена, стимулированной IFN-рецептором (44), и гибелью некроптозных клеток (27), казалось правдоподобным постулировать, что гомологичная RV-инфекция локально подавляет IFN-опосредованные воспалительные функции в тонкой кишке.Наши гистопатологические данные (рис. 3C) подтверждают, что на локальном уровне тонкой кишки мышиные детеныши, инфицированные EW RV, в значительной степени защищены от серьезного воспалительного повреждения из-за воздействия экзогенного эндотоксина. В связи с этими результатами возникает интересный вопрос, который мы в настоящее время изучаем: могут ли подобные защитные эффекты RV на воспаление проявляться вне кишечника. Гомологичная инфекция RV также блокирует программу экспрессии воспалительных генов, стимулированную рецептором IFN, на что указывает обнаружение кишечной секреции нескольких стимулированных IFN воспалительных цитокинов (RANTES (34–36), IP-10 (35, 37), Eotaxin (38) ), GRO-α (39, 40), MIP-1b (36, 41, 42)) были значительно ниже у мышиных RV-инфицированных детенышей по сравнению с ограниченным репликацией гетерологичного обезьяньего RV, несмотря на значительно более высокие уровни репликации штамм мышей (рис.3А).
Ротавирус эффективно разрушает несколько типов IFNR в инфицированных клетках (8), что, по прогнозам, напрямую ограничивает как IFN-опосредованную гибель воспалительных клеток, так и амплификацию гена за счет истощения поверхностной экспрессии IFNR. Белок (белки) RV, который опосредует деградацию IFNR, не был идентифицирован, хотя кодируемый RV неструктурный белок NSP1, по-видимому, является наиболее вероятным кандидатом из-за его способности ингибировать передачу сигналов STAT1 и вызывать деградацию как IRF3, так и bTrCP. (26, 32, 61).В этом исследовании мы исследовали, ингибируется ли функция IFNR в клетках-свидетелях также RV. Ранее мы сообщали, что штамм свиного RV (SB1-A) предотвращает фосфорилирование STAT1-Y701 в неинфицированных RV-свидетелях HT-29 клеток после относительно короткого (т.е. 30 минут) эктопического воздействия IFN (26). Предотвращают ли не свиные RV также передачу сигналов, опосредованную IFN, в клетках-свидетелях, не изучалось дополнительно с использованием измененных условий стимуляции IFN. Здесь, используя более длительную стимуляцию IFN (т.е. 6 h) в сочетании с анализом FACS, мы демонстрируем, что несколько штаммов не свиного RV, которые, как было показано ранее, опосредуют деградацию IFNR в инфицированных клетках, также индуцируют эффективную устойчивость к IFN-направленному STAT1-pY701 в клетках-свидетелях (фиг. 4A).
Способность различных штаммов RV блокировать передачу сигналов IFN в клетках-свидетелях является вторым направлением вирусной стратегии по подавлению противовирусных и воспалительных функций, опосредованных IFNR. Эффекторные функции различных IFN точно титруются чувствительным рецептор-центрированным модулирующим механизмом отрицательной обратной связи (62–64), гарантируя, что чрезмерное воспалительное повреждение хозяина не происходит из-за неконтролируемой передачи сигналов IFNR с прямой связью.Нарушение регуляции этого равновесия может привести либо к устойчивости к IFN, примером которой являются раковые клетки (65), либо к острым воспалительным патологиям (66), таким как сепсис. Ранее мы продемонстрировали, что парентеральное введение IFN γ приводит к эффективной активации STAT1-pY701 в эпителиальных клетках тонкого кишечника, процесс, который значительно подавляется во время гомологичной инфекции EW RV у грудных мышей (8). Аналогичное нарушение IFNR-опосредованной активации STAT1 мышиным EW RV происходило, если очищенный IFN-β или IFN- λ использовался для стимуляции их родственных рецепторов (8).Более ранние исследования также идентифицировали STAT1-независимый путь, который регулирует экспрессию гена, стимулированную IFN γ (67–70). Совсем недавно продукция воспалительных цитокинов в ответ на стимуляцию толл-подобных рецепторов (TLR) была значительно подавлена эктопической стимуляцией IFN γ и IFNβ стимуляцией STAT1-нулевых клеток путем перенаправления передачи сигналов IFNGR1 и IFNAR1 на ингибирование обратной связи (50 ). В этом исследовании мы изучили, может ли такая альтернативная передача сигналов IFNR также узурпироваться RV для широкого подавления врожденных противовирусных и воспалительных реакций.
Несколько транскриптов, активируемых во время фазы прямой амплификации IFNR, также могут быть эффективно индуцированы внутриклеточной дцРНК посредством передачи сигнала от различных рецепторов распознавания образов, включая RIG-I-подобные рецепторы (RLR), толл-подобные рецепторы (TLR). ) и дцРНК-зависимой протеинкиназы (PKR) (3). Поэтому мы сравнили способность стимуляции dsRNA и IFNβ к усилению экспрессии противовирусных и воспалительных генов во время инфицирования RV клеток HT-29 (рис.4Б). Поскольку инфицированные RV + клетки HT-29 эффективно истощены по различным IFNR, в нашем эксперименте предполагалось, что стимулированная IFNβ транскрипция происходит через IFNR, экспрессируемые на неинфицированных RV-свидетелях, хотя мы не продемонстрировали это напрямую. Результаты ясно указывают на эффективное ингибирование транскрипционных ответов с прямой связью на стимуляцию IFNR RV по сравнению со стимуляцией внутриклеточной дцРНК (фиг. 4B). Интересно, что стимуляция IFNβ во время инфекции RV подавляла транскрипцию HERC5 , IFNG , IL22, IRF8, IL29, и IL1B , что позволяет предположить, что стимуляция IFNAR1 (предположительно неинфицированных клеток-свидетелей RV) также может запускать режим ингибиторной обратной транскрипции (рис.4С).
В последующих экспериментах мы количественно оценили кишечную IFNR-стимулированную транскрипцию во время гомологичной EW RV (или ложной) инфекции путем эктопической стимуляции IFNGR1 у сосущих мышей (фиг. 5A). Эти результаты продемонстрировали, что, в отличие от режима транскрипции с прямой связью «прайминга» в псевдоконтроле, стимуляция IFN γ во время инфицирования EW RV потенцирует снижение ISG и воспалительных транскриптов ( STAT1, ISG15, IFI204, DHX58, DDX58, MX2, CCL5, TRAIL, NOS2, и CXCL10 ) (рис.5Б-С). Кроме того, мы обнаружили, что эктопическая стимуляция IFNGR1 во время инфицирования EW RV (но не стимуляция IFN γ или только EW RV) синергетически увеличивает транскрипцию противовоспалительных транскриптов (фиг. 5D). Интересно, что некоторые из этих генов с повышенной регуляцией (например, S100A8, BIRC5, ARG2, REG3G, и MKI67 ) являются мишенями сигнального пути STAT3, активация которого, как сообщается, подавляет опосредованные STAT1 воспалительные и противовирусные программы из-за стимуляции IFNR ( 33, 68–70).В настоящее время мы изучаем, происходит ли активация STAT3 во время инфекции RV и опосредует ли обратную связь IFNR.
В совокупности наши результаты демонстрируют, что RV эффективно перепрограммирует транскрипцию, передаваемую IFNR, в сторону подавления противовирусной и воспалительной амплификации (рис. 3, 4, 5), процесса, эффективность которого, как ожидается, будет увеличиваться за счет IFN, генерируемых в местной кишечной среде во время кишечной Инфекция правого желудочка (рис. 1). Способность RV-инфекции репрограммировать транскрипционную функцию IFNR определяет лежащий в основе механизм, с помощью которого RV также может контролировать другие функции IFNR, такие как опосредованная расщеплением каспазой гибель клеток.
МАТЕРИАЛЫ И МЕТОДЫ
Вирусы и реагенты
Штаммы RV, адаптированные к культуре тканей (обезьяний RRV, SA11-4F, SA11-5S, человеческий WI-61, свинья SB1-A, бычий UK), были размножены на африканских зеленых мартышках. клетки почек (MA104), как описано ранее (71). Титры вирусов определяли с помощью анализа бляшек в клетках MA104. Клетки HT-29 высевали в 6-луночные или 24-луночные кластерные планшеты, и полностью конфлюэнтные монослои инфицировали через 2-3 дня, как описано. Штамм EW RV мышей, не адаптированный к тканевой культуре, получали в виде кишечного гомогената от грудных мышей BALB / C, инфицированных RV.Дозу от диареи (DD 50 ) определяли, как описано ранее (16). Клетки HT29 кишечного эпителия человека и клетки HEK293 эмбриональных почек были приобретены из Американской коллекции типовых культур (ATCC) и сохранены в среде Advanced D-MEM Ham’s F12 (Cellgro) или в среде D-MEM (D-MEM, Cellgro), содержащей 10% фетальной сыворотки теленка. (Invitrogen) с добавлением заменимых аминокислот, глутамина, пенициллина и стрептомицина соответственно. Очищенный человеческий IFN-β (PBL), человеческий IFN-λ (PBL) и человеческий IFN-γ (Millipore) использовали для стимуляции клеток.Использовали очищенный без носителя IFN-β человека (PBL), IFN-γ мыши, TNF-α человека (R&D System, Миннеаполис, Миннесота), LPS (Sigma). Циклогексимид (Sigma) и высокомолекулярный LyoVec poly (I: C) (Invivogen) использовали в концентрации 10 мкг / мл и 2,5 мкг / мл соответственно.
Инфекция мышей и сбор тканей
Всех мышей содержали в отделении ветеринарной медицины VA Palo Alto Health Care System (VAPAHCS), и все эксперименты следовали протоколам, утвержденным Комитетом по уходу и использованию животных VAPAHCS.Детенышей Sv129 в возрасте 3-5 дней инфицировали через желудочный зонд 10 4 DD 50 мышиных EW RV, 10 7 БОЕ обезьяньих RRV или ложно инфицировали 1X средой M199 (Gibco), как описано (16 ). (16). В указанные моменты времени был удален тонкий кишечник. Срезы хранили при -80 ° C или фиксировали в 4% параформальдегиде для обработки и последующих анализов. Протокол изоляции IEC, как описано ранее (72), был немного изменен. Вкратце, кишечник удаляли и продольно открывали в среде RPMI с добавлением пенициллина, стрептомицина и L-глутамина (Gibco).Кишки промывали, разрезали на более мелкие кусочки и помещали в 1X PBS, содержащий 5 мМ EDTA и 5 мМ DTT (Sigma-Aldrich). На этом этапе объединяли ткани от 3-4 детенышей (на каждую инфекционную группу). Затем ткани помещали на шейкер на 10 минут при комнатной температуре. Высвободившиеся клетки затем фильтровали через фильтр с ячейками 100 мкм в свежую среду RPMI FBS (RPMI с добавлением 10% фетальной бычьей сыворотки, пенициллина, стрептомицина и L-глутамина) и сразу же помещали на лед. Оставшиеся ткани затем инкубировали со свежей средой RPMI FBS и встряхивали в течение дополнительных 10 минут с последующей последней промывкой в среде RPMI FBS в течение часа.Клетки фильтровали, промывали и ресуспендировали в PBS для дальнейших анализов.
Стимуляция мышей и анализы Luminex
Для эктопической стимуляции IFNGR1 мышей-сосунков, инфицированных 10 4 DD 50 мышиных EW RV (или ложно-инфицированных), внутрибрюшинно вводили очищенный мышиный IFN-γ (1 мкг IFN-γ на pup) или IFN-λ2 через 12 hpi и умерщвляют через 12 часов для анализа транскриптов с помощью qRT-PCR. Для стимуляции эндотоксином мышей 129sv в возрасте 3-5 дней перорально инокулировали через желудочный зонд 10 4 DD 50 мышиных RV-EW.При 3 dpi мышам внутрибрюшинно вводили PBS или очищенный LPS (Sigma, 10 мг / кг массы тела) и умерщвляли через 6 часов для анализа срезов ткани, фиксированных параформальдегидом и залитых парафином, путем окрашивания гематоксилином и эозином. Для измерения кишечных цитокинов собирали кишечник мышей и взвешенные объединенные ткани гомогенизировали в ледяном PBS, содержащем смесь ингибиторов протеазы и фосфатазы (Sigma), с использованием ручного микропеста (Thermo Fisher). Лизаты осветляли двумя последовательными раундами центрифугирования при 10,000X G в течение 10 минут и анализировали на уровни цитокинов в Центре мониторинга иммунитета человека в Стэнфордском университете.Наборы Mouse 38-plex были приобретены у eBiosciences / Affymetrix и использовались в соответствии с рекомендациями производителя. Планшеты считывали с помощью прибора Luminex 200 с нижней границей 50 шариков на образец на цитокин. Во все лунки добавляются специальные шарики для анализа от Radix Biosolutions.
Вестерн-блоттинг
Приготовление клеточных лизатов и иммуноблоттинг in vitro выполняли, как описано. Для образцов мышей IEC, обработанные EDTA / DTT, лизировали в буфере для анализа радиоиммунопреципитации (RIPA) с добавлением ингибиторов протеаз и фосфатаз (Roche).К каждому образцу добавляли равные объемы 2X буфера Лэммли (Sigma-Aldrich) и анализировали с помощью иммуноблоттинга. Блоты зондировали следующими первичными антителами: анти-каспаза 3, анти-каспаза 8, анти-каспаза 9, анти-расщепленная каспаза 3, анти-расщепленная каспаза 8, анти-расщепленная каспаза 7, анти-PARP, анти-GAPDH, анти-сурвивин, анти-STAT-3, анти-p-STAT-3 (Y705) (все от Cell Signaling Technologies) и капсидное антитело против RV (от Santa Cruz Biotechnologies). Затем блоты инкубировали с вторичными антителами против кролика или мыши, конъюгированными с пероксидазой хрена (HRP), инкубировали с реагентом для обнаружения ECL и экспонировали на пленке (GE Healthcare).Все денситометрические количественные определения белковых блотов анализировали с использованием программного обеспечения ImageJ для анализа и нормализовали по полосам белка GAPDH.
Транскрипционный анализ
Для in vi v o анализ транскриптов мышей умерщвляли и срезы тонкой кишки собирали на льду в Trizol (Life Technologies). Для исследований in vitro клетки сначала промывали PBS, а затем лизировали в Trizol. Тотальную РНК экстрагировали в соответствии с инструкциями производителя и подвергали расщеплению ДНКазой перед использованием в qRT-PCR.Синтез кДНК и последующая микрофлюидная ПЦР на платформе Fluidigm проводили, как описано ранее (73). Серийные 10-кратные разведения эталонной РНК мыши или человека (Agilent) проводили в двух экземплярах для каждого цикла ПЦР.
Анализ проточной цитометрии
Для анализа проточной цитометрии экспрессии каспаз и жизнеспособности клеток in vivo кишечник мыши собирали, как описано выше, и эквивалентное количество IEC, обработанных EDTA / DTT, окрашивали на жизнеспособность клеток с помощью Invitrogen’s LIVE / DEAD Fixable Набор Aqua Dead Cell Stain Kit в соответствии с протоколом производителя.Клетки блокировали очищенным крысиным антимышиным блоком CD16 / CD32 Fc (BD Biosciences) и окрашивали на следующие мышиные поверхностные маркеры: анти-CD45 (клон 30-F11) v450 (BD Biosciences), анти-CD326 (Ep-CAM / ESA, клон G8.8) PE (Biolegend) и анти-CD44 (клон IM7) PerCP / Cy5.5 (Biolegend), как описано ранее (7). Затем клетки фиксировали и повышали проницаемость с помощью буфера BD Cytofix / Cytoperm Fixation / Permeabilization в соответствии с протоколом производителя и впоследствии использовали для окрашивания внутриклеточных белков.Для окрашивания внутриклеточного белка клетки блокировали блоком BD Fc и инкубировали с неконъюгированными антителами к расщепленной каспазе 8 (Cell Signaling Technologies) или расщепленной каспазе 3 (Cell Signaling Technologies) вместе с конъюгированными антителами к VP6 RV (1E11-Texas Red) и Ki67 (Alexa Fluor 647, BD Biosciences). Затем клетки инкубировали с вторичным козьим антителом против кроличьего Alexa Fluor 488 (Molecular Probes) для обнаружения расщепленных каспаз. Все данные были получены с использованием LSRII (BD Biosciences) и проанализированы с помощью программного обеспечения FlowJo (Treestar Inc).Окрашивание и анализ проточной цитометрии клеток HT-29 выполняли, как описано.
TUNEL и анализы апоптоза
Для TUNEL после окрашивания поверхности эпителиальных клеток кишечника (как описано выше) клетки фиксировали и повышали проницаемость для последующего определения фрагментации ДНК с использованием набора Roche для определения гибели клеток in situ — флуоресцеин (согласно протоколу производителя. ). Вкратце, клетки фиксировали в 2% PFA и пермеабилизировали в 0,1% Triton X-100 в 0,1% натрийцитратном буфере и инкубировали с терминальной дезоксинуклеотидилтрансферазой (TdT) вместе с раствором для мечения флуоресцеин-dUTP в течение 1 часа при 37 ° C.В качестве положительного контроля проницаемые IECs обрабатывали ДНКазой I (Ambion) в течение 30 минут при 37 ° C (для индукции фрагментации ДНК) перед реакцией TUNEL. Также был включен дополнительный отрицательный контроль (реакции мечения проводились без фермента TdT). Включение меченого dUTP анализировали на проточном цитометре LSRII. Для анализа экспрессии маркера апоптоза в клетках HT-29 клетки промывали PBS, окрашивали Apopxin Green (разведение 1: 100, Abcam) при комнатной температуре в течение 10 минут и промывали перед исследованием с помощью флуоресцентной микроскопии.
Статистический анализ
Дисперсионный анализ (ANOVA) и двусторонний t-критерий Стьюдента были выполнены с использованием программного обеспечения GraphPad InSTAT. Подписи к рисункам указывают на конкретные использованные тесты, а значения P <0,05 считались значимыми. Все планки погрешностей представляют собой стандартную ошибку среднего.
Функциональное взаимодействие между интерферонами типа I и II необходимо для ограничения воспаления тканей, вызванного вирусом гриппа A
Abstract
Контроль хозяина вируса гриппа A (IAV) связан с обильным воспалением легких, характеризующимся притоком миелоидных клеток и производством провоспалительных цитокинов, включая интерфероны (IFN).Однако неясно, как иммунная система очищает вирус, не вызывая смертельной иммунопатологии. Здесь мы демонстрируем, что помимо известной противовирусной активности, передача сигналов STAT1 координирует воспаление хозяина во время инфекции IAV у мышей. Этот регуляторный механизм зависит от передачи сигналов рецептора IFN типа I и IFN-γ и, что важно, требует функционального взаимодействия между двумя путями. Защитная функция IFN типа I связана не только с рекрутированием классических воспалительных моноцитов Ly6C hi в IAV-инфицированные легкие, но также с предотвращением чрезмерной активации моноцитов IFN-γ.Неожиданно, IFN типа I предпочтительно регулируют передачу сигналов IFN-γ в популяциях мононуклеарных клеток Ly6C lo , а не воспалительных Ly6C hi . В отсутствие передачи сигналов IFN типа I моноциты / макрофаги Ly6C lo становятся фенотипически и функционально более провоспалительными, чем клетки Ly6C hi , что свидетельствует о непредвиденной функции субнабора мононуклеарных клеток Ly6C lo в тканевом воспалении. Кроме того, мы показываем, что IFN типа I используют разные механизмы для регуляции переноса моноцитов и нейтрофилов.Передача сигналов IFN типа I необходима, но недостаточна для предотвращения рекрутирования нейтрофилов в легкие IAV-инфицированных мышей. Вместо этого требуется взаимодействие IFN типа I и продуцируемого лимфоцитами IFN-γ, чтобы регулировать нейтрофильный ответ ткани на IAV. Наше исследование демонстрирует, что взаимодействие IFN связывает врожденный и адаптивный противовирусный иммунитет с управлением воспалением тканей и выявляет дополнительный уровень сложности для IFN-зависимых регуляторных механизмов, которые действуют для предотвращения чрезмерной иммунопатологии при сохранении антимикробных функций.
Сведения об авторе
Вирус гриппа A (IAV) является основной причиной респираторной инфекции и вызывает сильное острое воспаление, проявляющееся в привлечении моноцитов и нейтрофилов, а также в производстве провоспалительных цитокинов в инфицированных легких. Интерфероны (IFN) сильно индуцируются IAV и, как известно, опосредуют устойчивость хозяина к инфекции. Однако, в отличие от их хорошо изученного ингибирующего действия на репликацию вирусов, влияние IFN на воспалительные реакции хозяина менее изучено.В этой рукописи мы демонстрируем, что антивирусная передача сигналов IFN также необходима для организации тканевого ответа, связанного с защитой от инфекции IAV у мышей. Важно отметить, что мы идентифицируем, что IFN типа I перекрестно регулируют и взаимодействуют с IFN-γ, чтобы ингибировать активацию моноцитов и инфильтрацию нейтрофилов, соответственно. Это исследование также демонстрирует, что моноциты / макрофаги Ly6C lo потенциально могут опосредовать вызванное вирусом гриппа воспаление, предполагая, что IFNs диктуют гомеостаз по сравнению с воспалительной функцией мононуклеарных фагоцитов при вирусной инфекции.Наше исследование раскрывает новый IFN-зависимый регуляторный механизм, предназначенный для предотвращения чрезмерной иммунопатологии при сохранении его антимикробных функций. Более того, эти наблюдения имеют особое значение для понимания механизмов, лежащих в основе сильного воспалительного ответа, связанного с летальными штаммами IAV, и имеют значение для разработки новых иммунотерапевтических средств для лечения гриппа.
Образец цитирования: Stifter SA, Bhattacharyya N, Pillay R, Flórido M, Triccas JA, Britton WJ, et al.(2016) Функциональное взаимодействие между интерферонами типа I и II необходимо для ограничения воспаления тканей, вызванного вирусом гриппа А. PLoS Pathog 12 (1): e1005378. https://doi.org/10.1371/journal.ppat.1005378
Редактор: Пол Г. Томас, Детская исследовательская больница Св. Джуда, США
Поступила: 28 августа 2015 г .; Дата принятия: 9 декабря 2015 г .; Опубликовано: 5 января 2016 г.
Авторские права: © 2016 Stifter et al.Это статья в открытом доступе, распространяемая в соответствии с условиями лицензии Creative Commons Attribution License, которая разрешает неограниченное использование, распространение и воспроизведение на любом носителе при условии указания автора и источника
Доступность данных: Все соответствующие данные находятся в пределах документ и вспомогательные информационные файлы к нему.
Финансирование: Эта работа была поддержана Национальным советом по здравоохранению и медицинским исследованиям (NHMRC) Австралийского гранта проекта (APP1051742).WJB поддерживается грантом Центра передового опыта исследований в области борьбы с туберкулезом NHMRC (APP1043225). NB поддерживается Австралийской премией для аспирантов. Финансирующие организации не играли никакой роли в дизайне исследования, сборе и анализе данных, принятии решения о публикации или подготовке рукописи.
Конкурирующие интересы: Авторы заявили, что никаких конкурирующих интересов не существует.
Введение
Вирус гриппа A (IAV) является основной причиной респираторных инфекций и постоянной угрозой для здоровья людей во всем мире.Очистка хозяина от IAV, который инфицирует в первую очередь эпителиальные клетки дыхательных путей, требует развития как врожденных, так и адаптивных иммунных ответов [1,2]. Интересно, что недавние исследования показали, что иммунный ответ хозяина, а не цитопатический эффект вирусной инфекции, играет ключевую роль в развитии тканевой патологии и смертности хозяина [3-5].
IAV вызывает острое воспаление легких, связанное с привлечением воспалительных моноцитов и нейтрофилов в инфицированных легких (см. Обзор [6]).Хотя очевидно, что повышенное накопление нейтрофилов в инфицированных легких связано с повышенной смертностью после инфекции IAV [7,8], рекрутирование моноцитов может быть защитным или пагубным [9,10], предполагая, что моноциты могут играть многофакторную роль в инфекции. . Современное понимание моноцитов предполагает, что существует по крайней мере два основных подмножества: классические моноциты Ly6C hi и неклассические моноциты Ly6C lo [11]. Известно, что классические моноциты Ly6C hi опосредуют различные воспалительные состояния [12] и накапливаются в больших количествах в легких, инфицированных IAV [13].Напротив, неклассические клетки Ly6C lo , как было показано, «патрулируют» сосудистую сеть, чтобы очистить поврежденные эндотелиальные клетки и внести вклад в ремоделирование ткани во время фазы разрешения воспаления [14]. Интересно, что патрулирующие моноциты Ly6C lo , как было показано, дифференцируются в альтернативно активированные макрофаги во время инфекции Listeria monocytogenes [14]. Однако фенотип и функция моноцитов / макрофагов Ly6C lo (Mo / Mϕ) при инфекции IAV в настоящее время неизвестны.
Помимо набора миелоидных клеток, IAV индуцирует продукцию провоспалительных цитокинов, включая интерфероны I и II типа (IFN), у инфицированных животных. Семейство IFN типа I состоит из ~ 20 различных членов, которые считаются важными для противовирусного и противоракового иммунитета [15], тогда как единственный член IFN типа II, IFN-γ, играет важную роль в активации Mo / Mϕ и защите против внутриклеточных бактериальных и паразитарных инфекций (см. обзор [16]). В отличие от его широко изученной функции в инициировании клеточно-автономного антивирусного состояния, механизмы, с помощью которых передача сигналов IFN регулирует ответы тканей хозяина на инфекцию IAV, плохо изучены.Несколько недавних исследований показали, что индуцированные вирусом IFN типа I способствуют накоплению классических моноцитов Ly6C hi в дыхательных путях и легких мышей, инфицированных IAV [17,18]. Однако неясно, регулируют ли эти цитокины также функцию легочных моноцитов в отношении устойчивости к инфекции IAV. Более того, вклад IFN-γ в индуцированное IAV воспаление легочной ткани четко не определен.
Мы сообщаем в этом исследовании, что передача сигналов IFN и IFN-γ типа I играет плейотропную роль в воспалительной реакции легких на IAV.Важно отметить, что перекрестная регуляция и взаимодействие IFN важны для подавления воспаления тканей, вызванного моноцитами и нейтрофилами. Противодействуя передаче сигналов IFN-γ для ингибирования активации Mo / Mϕ, IFN типа I действуют синергично с IFN-γ, подавляя инфильтрацию нейтрофилов. Более того, мы демонстрируем, что в отсутствие передачи сигналов IFN типа I Ly6C lo Mo / Mϕ становится более провоспалительным, чем их аналоги Ly6C hi . Поскольку популяция Ly6C lo Mo / Mϕ традиционно связана с ремоделированием ткани, а не с воспалением, наши результаты также показывают нераспознанный провоспалительный потенциал Ly6C lo Mo / Mϕ и предполагают, что IFNs диктуют гомеостаз по сравнению с воспалительной функцией мононуклеаров. клетки при вирусной инфекции.
Результаты
Передача сигналов STAT1 контролирует воспалительную реакцию легких на инфекцию IAV
Сублетальная интраназальная (in) инфекция вирусом гриппа A / Puerto Rico / 8 (PR8) у мышей дикого типа (WT) характеризуется прогрессирующей потерей веса, достигающей пика на 10-й день, после чего мыши выздоравливают и очищаются от вируса. инфекции (рис. 1А и [19]). Чтобы исследовать функцию IFNs в формировании ответа хозяина на инфекцию IAV, мы сначала исследовали экспрессию гена IFN и наблюдали, что у мышей WT типа I IFN Ifna и Ifnb быстро повышались на 3-й день после инфицирования. (п.i.), единственный IFN типа II, Ifng , значимо не индуцировался до 7 дня p.i. (Рис. 1B). Экспрессия ИФН типа I и II снижалась до исходного уровня к 10-му дню пи. У мышей WT клеточный иммунный ответ на инфекцию IAV характеризовался сильным притоком моноцитов, который достигал пика на 7-й день и затем снижался к 10-му дню, т.е. В результате Т-клетки стали доминирующей субпопуляцией иммунных клеток в легких (рис. 1С). В отличие от контроля WT, мыши Stat1 с дефицитом , лишенные передачи сигналов как IFN, так и IFN-γ типа I, продемонстрировали клеточный ответ, обогащенный нейтрофилами (рис. 1D), что подчеркивает критическую важность передачи сигналов IFN в организации реакции защитной ткани на Инфекция IAV.Затем мы количественно оценили вирусную нагрузку на 3-й и 7-й дни p.i. с использованием как qRT-PCR, так и анализа классического образования бляшек (PFU). В то время как количество копий мРНК вирусного нуклеопротеина (NP), определенное с помощью qRT-PCR, было сопоставимым у мышей WT и Stat1 — / — , последние животные показали незначительное увеличение (0,4 log) БОЕ на 7 день (рис. 1E). . Степень дефекта вирусного контроля у мышей Stat1 — / — согласуется с данными, о которых сообщалось ранее [20,21], предполагая, что нарушение регуляции тканевого ответа у мышей Stat1 — / — невозможно полностью объяснить. повышением вирусной нагрузки.
Рис. 1. Передача сигналов STAT1 диктует воспалительный ответ клеток хозяина на IAV независимо от регуляции миелопоэза.
(A) Изменение массы тела после инфицирования IAV. Мыши были инфицированы i.n. с IAV PR8 (20 БОЕ) и ежедневным контролем массы тела. Представленные данные представляют собой средний вес ± стандартное отклонение и представляют 2 независимых эксперимента (5 мышей / эксперимент). (B) Экспрессию мРНК Ifna , Ifnb и Ifng в легких инфицированных мышей анализировали с помощью qRT-PCR в указанные моменты времени.Представленные данные представляют собой среднее кратное увеличение по сравнению с неинфицированными контролями. Данные были объединены из 2 независимых экспериментов с аналогичной тенденцией (n = 5–9 мышей на момент времени). Кружки обозначают отдельных мышей, а столбцы — средние по группе. (C) Доля лейкоцитов легких после инфекции IAV. Показанные данные представляют собой среднее значение ± стандартное отклонение (n = 4) и представляют не менее 3 независимых экспериментов. (D) Вертикальные полосы на срезах показывают относительные пропорции основных субпопуляций лейкоцитов легких, количественно определенные с использованием проточного цитометрического анализа у инфицированных мышей WT и Stat1 — / — на d7 p.i .. Представленные данные являются средними для трех мышей и представляют 3 независимых эксперимента. (E) Общее количество копий мРНК NP IAV и БОЕ в легких инфицированных мышей при d3 (светлые столбцы) и d7 (темные столбцы) p.i. как определено с помощью qRT-PCR и анализа бляшек MDCK, соответственно. Показанные данные представляют собой среднее число копий ± стандартное отклонение или среднее значение БОЕ ± стандартное отклонение и были объединены из 3 независимых экспериментов (n = 3–11 мышей на группу). (F и G) Репрезентативные графики проточной цитометрии и сводные данные, отображающие пропорции (F) гемопоэтических стволов / предшественников (Lineage — CD117 + ), а также (G) зрелых моноцитов (Mono, CD11b + Ly6C + Ly6G — ) и нейтрофилы (Neut, CD11b + Ly6G + Ly6C — ) в BM инфицированных WT и Stat1 — / — мышей на d7 p .i .. Кружки обозначают отдельных мышей, а столбцы — средние по группе. Данные были объединены из 2 независимых экспериментов (n = 4–6 мышей).
https://doi.org/10.1371/journal.ppat.1005378.g001
Чтобы определить, является ли измененное воспаление легких у инфицированных мышей Stat1 — / — результатом нарушения регуляции миелопоэза в костном мозге (BM) или клеточного транспорта на периферии или в обоих, мы проанализировали гемопоэтические клетки в BM инфицированных мышей WT и Stat1 — / — с помощью проточной цитометрии.Популяции стволовых / предшественников клеток обычно определяют как клетки Sca1 + cKit + (CD117 + ) (LSK), отрицательные по клону [22,23]. Однако, поскольку экспрессия Sca1 регулируется IFN (S1 фиг. И [22]), мы не включили окрашивание Sca1 в анализ. Мы не наблюдали существенной разницы в процентном отношении клон-отрицательных клеток-предшественников CD117 + (рис. 1F) или зрелых находящихся в BM CD11b + Ly6G + Ly6C lo нейтрофилов и CD11b + Ly6G — Ly6C hi моноцитов (рис. 1G) у инфицированных мышей WT и Stat1 — / — , что позволяет предположить, что передача сигналов IFN играет минимальную роль в регуляции центрального миелопоэза в костном мозге во время инфекции IAV.
Внутренняя передача сигналов IFN типа I способствует рекрутированию воспалительных моноцитов Ly6C
hi в легкие, инфицированные IAVЧтобы определить относительный вклад IFN типа I и II в рекрутирование моноцитов при IAV-инфекции, мы проанализировали популяции моноцитов в легких инфицированных WT, Ifnar1 — / — , Ifngr1 — / — и Stat1 — / — мышей с использованием проточной цитометрии.По сравнению с мышами WT, мыши Ifnar1 — / — и Stat1 — / — показали значительное снижение моноцитов CD11b + Ly6C hi в легких на 7 день p.i. (Рис. 2A, 2B и 2C). Разница маловероятна из-за кинетических вариаций легочного ответа на IAV у мышей с достаточной и недостаточной передачей сигналов IFN, поскольку значительный приток лейкоцитов в инфицированные легкие не наблюдался до 7 дня p.i. во всех группах (рис. 2В и 2С).В отличие от Ifnar1 — / — животных, рекрутирование моноцитов у мышей Ifngr1 — / — в значительной степени не затрагивалось, что позволяет предположить, что одной передачи сигналов IFN типа I достаточно для запуска рекрутирования моноцитов в инфицированные легкие. Затем мы определили, действуют ли IFN типа I прямо или косвенно на популяции моноцитов, регулируя их доставку в легкие. Смертельно облученных мышей-реципиентов CD45.1 + WT восстанавливали равным количеством CD45.1 + WT и CD45.2 + Ifnar1 — / — КМ клетки. После полного восстановления (8–10 недель) мышей-химер инфицировали IAV и анализировали клеточный ответ на 7 день p.i. (Рис 2D). Мы обнаружили, что дефект рекрутирования воспалительных моноцитов Ly6C hi , наблюдаемый у мышей Ifnar1 — / — , не восстанавливался в CD45.2 + Ifnar1 — / — клеток в смешанные Ifnar1 — / — и Ifnar1 + / + BM химерные мыши (рис. 2E).Таким образом, соотношение моноцитов Ly6C hi / Ly6C lo в компартменте CD45.2 + Ifnar1 — / — было значительно ниже, чем в компартменте CD45.1 + Ifnar1. + / + отсек, что указывает на то, что прямая передача сигналов IFN типа I в моноцитах Ly6C hi необходима для их рекрутирования в инфицированные легкие (рис. 2F).
Рис. 2. IFN типа I передают сигнал непосредственно в моноциты, способствуя их миграции в легкие, инфицированные IAV.
Мышей инфицировали IAV, и анализировали суспензии единичных клеток легких через день 7 p.i. методом проточной цитометрии. Показанные данные проточной цитометрии привязаны к популяциям клеток CD4 — CD8 — B220 — NK1.1 — Ly6G — SiglecF — . (A) Типичные графики проточной цитометрии, отображающие процентное содержание Ly6C hi и Ly6C lo Mo / Mϕ в легких инфицированных WT, Ifnar1 — / — , Ifngr1 — / — или Stat1 — / — мыши. (B) Сводные данные, показывающие процент моноцитов Ly6C hi в инфицированных легких. Данные были объединены по крайней мере из 3 независимых экспериментов (n = 6–10 мышей / группа). (C) Сводные данные, показывающие общее количество популяций клеток Ly6C hi и Ly6C lo в инфицированных легких на d3 (красные кружки) и d7 (черные кружки) pi. Данные были объединены по крайней мере из 3 независимых экспериментов ( n = 6–10 мышей / группа). (D) Схематическое изображение стратегии, используемой для создания, инфицирования и анализа смешанных химерных мышей BM. (E) Типичные графики проточной цитометрии, демонстрирующие процентное содержание Ly6C hi и Ly6C lo Mo / Mϕ в легких в CD45.1 + (WT) и CD45.2 + ( Ifnar1 — / — ) компартменты инфицированных смешанных мышей-химер BM. (F) Сводный анализ отношения подмножеств Ly6C hi к Ly6C lo в компартментах CD45.1 + (WT) и CD45.2 + ( Ifnar1 — / — ).Данные представляют 4 независимых эксперимента (n = 7–9 мышей). В B , C и F кружки обозначают отдельных мышей, а столбцы — средние по группе.
https://doi.org/10.1371/journal.ppat.1005378.g002
IFN типа I регулируют фенотип и функцию легочных моноцитов при IAV-инфекции
Затем мы исследовали, регулирует ли передача сигналов IFN типа I фенотип и функцию основных популяций моноцитов CD11b + в IAV-инфицированных легких.В соответствии с современными знаниями о субпопуляциях мононуклеарных фагоцитов [11], мы наблюдали, что моноциты Ly6C hi в IAV-инфицированных легких демонстрируют более высокую экспрессию CCR2, чем их аналоги Ly6C lo у мышей WT и Ifnar1 — / — мышей (рис. 3А). Более того, экспрессия интегрина LFA-1 (CD11a), ключевой молекулы, ответственной за функцию моноцитов [24], не изменилась ни на Ly6C hi , ни на Ly6C lo клетках, независимо от передачи сигналов IFN I типа.Поразительно, однако, что потеря передачи сигналов IFN типа I в популяциях Ly6C lo , но не Ly6C hi привела к значительному увеличению экспрессии MHC-II (IA), CD11c, CD16 / 32 и CD64, что позволяет предположить, что в Ifnar1 — / — мыши, Ly6C lo Mo / Mϕ проявляют фенотип, характерный для активированных провоспалительных моноцитов. В соответствии с этой гипотезой мы обнаружили, что Nos2 , известная провоспалительная молекула, продуцируемая преимущественно моноцитами, была более высоко экспрессирована в легких Ifnar1 — / — мышей, чем животных WT (рис. 3B), несмотря на то, что снижение уровня моноцитов Ly6C hi у мышей Ifnar1 — / — (рис. 2В).
Рис. 3. IFN типа I регулируют фенотип и эффекторные функции Mo / Mϕ в легких, инфицированных IAV.
(A) Типичные потоковые диаграммы, демонстрирующие популяции мононуклеарных фагоцитов Ly6C hi или Ly6C lo , и гистограммы, изображающие экспрессию поверхностных молекул на Ly6C hi или Ly6C lo клеточных субпопуляциях в легких зараженных d7 WT и Ifnar1 — / — мышей (n = 7 мышей / генотип). Показанные данные проточной цитометрии привязаны к CD11b + CD4 — CD8 — B220 — NK1.1 — Ly6G — SiglecF — популяции клеток. (B) Экспрессия Nos2 в легких инфицированных мышей, определенная на d7 p.i. с использованием qRT-PCR. Показанные данные представляют собой средние кратные увеличения (по сравнению с неинфицированными контролями) ± стандартное отклонение. Данные были объединены из 3 независимых экспериментов (n = 6–9 мышей / генотип). (C) Типичные графики потока внутриклеточной экспрессии NOS2 в легких инфицированных мышей WT и Ifnar1 — / — при d7 p.i .. Показанные данные проточной цитометрии привязаны к живым синглетным клеткам.Графики потока представляют 3 независимых эксперимента с n = 6-9 мышей / генотип. (D) Процент и количество NOS2-положительных мононуклеарных клеток в легких инфицированных мышей WT и Ifnar1 — / — мышей количественно определено с помощью проточной цитометрии. Данные были объединены из 3 независимых экспериментов с аналогичной тенденцией и представляют собой средние уровни ± стандартное отклонение (n = 6–9 мышей / генотип). (E) Типичные диаграммы потока, изображающие NOS2, присутствующую в клетках Ly6C hi и Ly6C lo .Показанные данные проточной цитометрии привязаны к клеточным популяциям CD11b + CD4 — CD8 — B220 — NK1.1 — Ly6G — SiglecF — . (F) Парный анализ процента и средней интенсивности флуоресценции (MFI) NOS2 в популяциях Ly6C hi и Ly6C lo в WT и Ifnar1 — / — мышей на d7 пи. Данные были объединены с 3 независимые эксперименты, и каждый набор парных данных представляет отдельную мышь.Статистический анализ проводили с использованием парного теста Стьюдента t .
https://doi.org/10.1371/journal.ppat.1005378.g003
В соответствии с анализом гена Nos2 , проточно-цитометрический анализ показал, что в легких инфицированных Ifnar1 наблюдалось значительное увеличение количества клеток, экспрессирующих NOS2. — / — мышей по сравнению с животными WT (рис. 3C). Дальнейший анализ показал, что множественные мононуклеарные клетки в легких, инфицированных IAV, продуцируют NOS2 (рис. 3D).Однако, в то время как моноциты Ly6C hi были основными клетками, продуцирующими NOS2 у животных WT, субпопуляции Ly6C hi и Ly6C lo вносили вклад в продукцию NOS2 в легких инфицированных Ifnar1 — / — мышей (Фиг.3D и 3E). Парный анализ двух субпопуляций моноцитов среди отдельных мышей показал, что у животных Ifnar1 — / — как процент клеток, экспрессирующих NOS2, так и общее количество продуцируемого NOS2 на клетку было выше у Ly6C lo . чем моноциты Ly6C hi , предполагая, что первая подгруппа более восприимчива к IFN-зависимой супрессии I типа (рис. 3F).Следовательно, IFN типа I способны регулировать не только транспорт, но также фенотип и эффекторную функцию мононуклеарных клеток в легких во время инфекции IAV.
IFN типа I подавляют IFN-γ-зависимую активацию моноцитов путем подавления рецептора IFN-γ
Поскольку исследованные выше поверхностные маркеры и NOS2, как известно, очень чувствительны к индукции IFN-γ [25–27], мы подозревали, что IFN-γ может играть роль в повышении регуляции этих молекул в инфицированных Ifnar1. — / — мыши.Действительно, на 3 день после рождения, момент времени, когда продуцируются минимальные уровни IFN-γ (рис. 1B), не было различий в экспрессии IA, CD11c или CD64 на клетках Ly6C hi и Ly6C lo ( S2 Рис). Чтобы напрямую продемонстрировать, что IFN-γ отвечает за активацию молекул, мы сравнили экспрессию IA и NOS2 в Ly6C lo Mo / Mϕ WT, Ifnar1 — / — , Ifngr1 — / — и Stat1 — / — мышей на 7 день после инфицирования IAV.Мы обнаружили, что повышенная экспрессия IA и NOS2, наблюдаемая в легких у животных Ifnar1 — / — , полностью исчезла у мышей Ifngr1 — / — , а также у мышей Stat 1 — / — у мышей, у которых отсутствуют сигнальные пути IFN типа I и IFN-γ (рис. 4A и 4B), что позволяет предположить, что повышающая регуляция IA и NOS2 в отсутствие передачи сигналов IFN типа I управляется IFN-γ.
Рис. 4. IFN типа I защищают Ly6C lo Mo / Mϕ от IFN-γ-индуцированной активации путем подавления рецептора IFN-γ.
(A) Типичные графики проточной цитометрии и (B) сводная статистика экспрессии I-A и NOS2 в Ly6C lo Mo / Mϕ. Показанные данные проточной цитометрии привязаны к клеточным популяциям CD11b + CD4 — CD8 — B220 — NK1.1 — Ly6G — SiglecF — . Кружки в (B) обозначают отдельных мышей, а столбцы представляют средние значения для группы. Данные были объединены из 3 независимых экспериментов с общим количеством 4–9 мышей в группе. (C) Типичные диаграммы потока, показывающие процентное содержание NOS2 и IA положительных CD45.1 + (WT) и CD45.2 + ( Ifnar1 — / — ) Ly6C lo Mo / Mϕ инфицированных смешанных мышей-химер BM в d7 пи.Показанные данные проточной цитометрии привязаны к CD11b + CD4 — CD8 — B220 — NK1.1 — Ly6G — SiglecF — популяциям клеток. Смешанные химерные мыши BM были созданы, инфицированы и проанализированы, как описано на фиг. 2D.Данные представляют 2 независимых эксперимента (n = 5). (D) Ifng и Ifngr1 экспрессия мРНК в легких инфицированных мышей на d7 p.i. были проанализированы с помощью qRT-PCR. Показанные данные представляют собой средние кратные увеличения (по сравнению с неинфицированными контролями) ± стандартное отклонение. Данные были объединены из 3 независимых экспериментов (n = 6–9 мышей / генотип). (E) Типичная проточная цитометрическая гистограмма экспрессии IFN-γ рецептора 1 (CD119) на Ly6C lo Mo / Mϕ и (F) средняя интенсивность флуоресценции экспрессии CD119 (± SD) на Ly6C lo Mo / Mϕ инфицированных мышей WT и Ifnar1 — / — мышей на d7 p.i .. Данные были объединены из 2 независимых экспериментов с участием 5–6 мышей на каждый генотип.
https://doi.org/10.1371/journal.ppat.1005378.g004
Чтобы определить, действуют ли интерфероны I типа непосредственно на моноциты, оказывая подавляющее действие, мы проанализировали продукцию NOS2 в смешанных, инфицированных IAV Ifnar1 + / + и Ifnar1 — / — Химерные мыши BM и обнаружили, что IFN типа I передают сигнал непосредственно на подавление продукции NOS2 как в Ly6C lo (рис. 4C), так и в Ly6C hi (рис. S3A). мононуклеарные клетки.Более того, как мы наблюдали ранее у мышей Ifnar1 — / — (рис. 3F), Ly6C lo Mo / Mϕ у химерных мышей BM экспрессировали более высокие уровни NOS2, чем их аналоги Ly6C hi (рис. S3B). , подтверждая, что повышенная чувствительность к ингибированию IFN типа I присуща этой подгруппе Mo / Mϕ. Затем мы определили, был ли усиленный IFN-γ-индуцируемый ответ у мышей Ifnar1 — / — , инфицированных IAV, следствием повышенной продукции или передачи сигналов IFN-γ, путем исследования экспрессии Ifng и Ifngr1 в инфицированных легких.Мы обнаружили, что, хотя экспрессия Ifng была сопоставима в легких инфицированных мышей WT и Ifnar1 — / — мышей, уровни Ifngr1 были значительно выше у Ifnar1 — / — животных (рис. 4D). , предполагая, что усиленная передача сигналов IFN-γ, а не продукция IFN-γ, ответственна за усиление IFN-γ-индуцируемого ответа у мышей Ifnar1 — / — . Чтобы проверить эту гипотезу, мы проанализировали экспрессию на клеточной поверхности рецептора 1 IFN-γ (CD119) с помощью проточной цитометрии и обнаружили, что экспрессия CD119 была значительно увеличена на Ly6C lo Mo / Mϕ мышей Ifnar1 — / — по сравнению с мышами. Мыши WT (рис. 4E и 4F).Следовательно, внутренняя регуляция уровней рецептора IFN-γ с помощью IFN типа I, по-видимому, является механизмом, с помощью которого активация Mo / Mϕ подавляется в присутствии IFN типа I.
Передача сигналов IFN типа I необходима, но недостаточна для ингибирования миграции нейтрофилов в легкие мышей, инфицированных IAV
Наши данные выше предполагают, что передача сигналов IFN типа I доминирует над IFN-γ в рекрутировании моноцитов Ly6C hi в легкие IAV-инфицированных мышей (рис. 2).Чтобы исследовать относительный вклад IFN типа I и типа II в STAT1-зависимое подавление миграции нейтрофилов (рис. 1D), WT, Ifnar1 — / — , Ifngr1 — / — и Stat1 — / — мышей инфицировали IAV, и относительное количество нейтрофилов в каждой линии мышей определяли с помощью проточной цитометрии. Интересно, что мы наблюдали, что как процент, так и количество нейтрофилов CD11b + Ly6G + были значительно повышены в легких Ifnar1 — / — , а также Ifngr1 — / — мышей. по сравнению с животными WT на 7 день, но не на 3 день p.я. (Рис. 5A, 5B и 5C). Важно отметить, что этот дефект был дополнительно преувеличен у мышей Stat1 — / — , что указывает на синергетическую функцию IFN типа I и II в подавлении рекрутирования нейтрофилов во время инфекции гриппа. Более того, в отличие от внутренней функции клеток IFN типа I в миграции моноцитов, описанной выше (рис. 2D), мы обнаружили, что процент нейтрофилов CD11b + Ly6C lo Ly6G + среди CD45.1 сопоставим. + WT и CD45.2 + Ifnar1 — / — отсеков до (S4, фиг.) И после (фиг. 5D) инфекции, что позволяет предположить, что IFN типа I регулируют миграцию нейтрофилов внешними клетками. Таким образом, IFN типа I используют различные механизмы для регуляции переноса моноцитов и нейтрофилов при IAV-инфекции и действуют совместно с IFN-γ для предотвращения накопления повреждающих ткань нейтрофилов в месте инфекции. Чтобы исследовать потенциальные внешние клеточные механизмы, ответственные за повышенное накопление нейтрофилов, мы измерили экспрессию генов известного привлекающего нейтрофилы хемокина Cxcl1 и цитокина Il1b у животных с дефицитом сигналов WT и IFN. Stat1 — / — мыши показали значительно более высокую экспрессию Cxcl1 и Il1b , чем в Ifnar1 — / — или Ifngr1 54 — / — Fig. 5E), что согласуется с мнением о том, что IFNs синергетически подавляют продукцию хемотаксических хемокинов / цитокинов нейтрофилами при IAV-инфекции.
Рис. 5. Передача сигналов IFN и IFN-γ типа I действует синергетически, подавляя перенос нейтрофилов в легкие, инфицированные IAV.
(A) Репрезентативные графики потока, изображающие популяции нейтрофилов Ly6G + легких. Показанные данные проточной цитометрии привязаны к популяциям клеток CD11b + CD4 — CD8 — B220 — NK1.1 — SiglecF — . (B) Процент нейтрофилов в мышах WT, Ifnar1 — / — , Ifngr1 — / — или Stat1 — / — , определяемый методом проточной цитометрии на d7 p.i .. (C) Число нейтрофилов в WT, Ifnar1 — / — , Ifngr1 — / — или Stat1 — / — мышей, определенных методом проточной цитометрии в d3 (красный кружки) и d7 (черные кружки) пи. Данные были объединены из 2 независимых экспериментов. (D) Процент нейтрофилов Ly6G + в крови и легких инфицированных смешанных химерных мышей BM. Смешанные химерные мыши CD45.1 + (WT) и CD45.2 + ( Ifnar1 — / — ) BM были созданы, инфицированы и проанализированы с использованием проточной цитометрии, как описано на фиг. 2D.Данные показывают процент нейтрофилов Ly6G + среди клеток CD45.1 + (WT) или CD45.2 + ( Ifnar1 — / — ) в крови или легких инфицированных смешанных химерных мышей BM. на d7 пи. Данные представляют 2 независимых эксперимента (n = 10–14 мышей / исследование). (E) Экспрессия мРНК Cxcl1 и Il1b в легких, проанализированная с помощью qRT-PCR при d7 p.i. Показанные данные представляют собой средние кратные увеличения (относительно неинфицированных контролей) ± стандартное отклонение.Данные были объединены из 3 независимых экспериментов с аналогичной тенденцией. В B , C и D кружки обозначают отдельных мышей, а столбцы — средние по группе.
https://doi.org/10.1371/journal.ppat.1005378.g005
Адаптивный IFN-γ и IFN типа I взаимодействуют для подавления переноса нейтрофилов в легкие мышей, инфицированных IAV
STAT1 является одним из многих факторов транскрипции, которые могут передавать сигналы рецептора IFN и IFN-γ типа I, но он также может опосредовать IFN-независимые функции [28–30].Следовательно, возможно, что повышенный приток нейтрофилов у мышей Stat1 — / — , инфицированных IAV, по сравнению с мышами Ifnar1 — / — , не зависит от передачи сигналов IFN-γ. Более того, неясно, требует ли IFN-γ-зависимое подавление нейтрофилов интактной передачи сигналов IFN I типа. IFN типа I, как известно, регулируют рекрутирование нейтрофилов путем передачи сигналов непосредственно в воспалительные моноциты, чтобы подавить их продукцию хемоаттрактивного хемокина нейтрофилов Cxcl2 [31].Чтобы ответить на эти вопросы, мы сгенерировали Ifnar1 — / — BM реконструированных химерных мышей WT, у которых отсутствует передача сигналов IFN типа I только в гематопоэтических клетках. Подобно мышам Ifnar1 — / — , эти химерные мыши также демонстрировали дефектное накопление моноцитов Ly6C hi в легких после инфекции IAV (S5 фиг.). Ifnar1 — / — Химерные мыши BM, обработанные антителом против IFN-γ на 2 и 6 день p.я. показали заметное снижение экспрессии I-A на моноцитах при анализе на 7 день (фиг. 6A). Это согласуется с нашим предыдущим наблюдением, что IFN-γ увеличивает экспрессию MHC класса II (фиг. 4), и подтверждает, что введение mAb успешно блокирует активность IFN-γ. Важно отметить, что в соответствии с результатами, представленными на фиг. 5, мы обнаружили, что как пропорция, так и количество нейтрофилов были значительно повышены у мышей, получавших анти-IFN-γ, по сравнению с необработанными животными (фиг. 6B). Мы также подсчитали нейтрофилы в бронхоальвеолярном лаваже (БАЛ) и наблюдали аналогичное увеличение количества нейтрофилов в дыхательных путях мышей, получавших анти-IFN-γ, по сравнению с необработанным контролем (рис. 6C), что указывает на то, что IFN-γ регулирует миграцию нейтрофилов в оба. паренхима легких и бронхоавеолярные воздушные пространства не зависят от передачи сигналов IFN типа I в воспалительных моноцитах.Эти результаты предполагают, что IFN-γ, продуцируемый адаптивным иммунным ответом на инфекцию IAV, регулирует воспалительный ответ ткани в дополнение к его противовирусной активности [1]. Чтобы проверить эту гипотезу, мы заразили мышей WT и Rag2 — / — , у которых отсутствуют В- и Т-клетки, и проанализировали IFN-γ и ответ нейтрофилов на d7 pi. Как и ожидалось, Rag2 — / — Мыши показали значительно сниженную экспрессию IFN-γ (фиг. 6D) и увеличенное накопление нейтрофилов Ly6G + в легких по сравнению с животными дикого типа (фиг. 6E).Вместе наши результаты раскрывают механизм, с помощью которого IFNs связывают врожденную и адаптивную иммунные системы, чтобы управлять легочным воспалением для устойчивости к инфекции IAV.
Рис. 6. Передача сигналов IFN-γ ингибирует перенос нейтрофилов в легкие, инфицированные IAV, независимо от IFN типа I и воспалительных моноцитов.
(A – C) Летально облученные мыши CD45.1 + WT были восстановлены клетками ВМ из мышей CD45.2 + Ifnar1 — / — и инфицированы IAV.Мышам вводили внутрибрюшинно. с 500 мкг нейтрализующего mAb против IFN-γ на d2 и d6 инфекции IAV или оставленные без лечения. Ткани легких и суспензии отдельных клеток анализировали при d7 p.i. с помощью проточной цитометрии. (A) Процент IA + Mo / Mϕ, (B и C) Процент и общее количество нейтрофилов Ly6G + в (B) легких и (C) BAL необработанных или антибактериальных -IFN-γ mAb, обработанные химерными мышами. Данные представляют 2 независимых эксперимента.Кружки обозначают отдельных мышей, а столбцы — средние по группе. (D – F) WT и Rag2 — / — мышей инфицировали IAV и проанализировали d7 pi. (D) экспрессию мРНК Ifng в легких анализировали с помощью qRT-PCR на d7 pi. Показанные данные представляют собой средние кратные увеличения (по сравнению с неинфицированными контролями) ± стандартное отклонение. Данные были объединены из 3 независимых экспериментов (n = 6-7 мышей / генотип). (E) Типичные графики проточной цитометрии и сводные данные, изображающие процентное содержание нейтрофилов Ly6G + в легких IAV-инфицированных WT и Rag2 — / — мышей d7 p.я. Данные были объединены из 2 независимых экспериментов с участием 5-8 мышей на каждый генотип. Кружки обозначают отдельных мышей, а столбцы — средние по группе. Показанные данные проточной цитометрии привязаны к популяциям клеток CD11b + CD4 — CD8 — B220 — NK1.1 — SiglecF — . (F) Схематическая диаграмма, иллюстрирующая функциональные точки пересечения между сигнальными путями IFN типа I и IFN-γ при инфекции IAV. IFN типа I взаимодействуют с IFN-γ, подавляя накопление нейтрофилов в месте инфекции (красные линии).IFN типа I противодействуют передаче сигналов IFN-γ, подавляя активацию Mo / Mϕ (черные линии).
https://doi.org/10.1371/journal.ppat.1005378.g006
Обсуждение
Хотя установлено, что IFN-опосредованная устойчивость к вирусной инфекции in vitro зависит от ингибирования репликации вируса [32], механизмы, с помощью которых IFNs защищают от инфекции in vivo , менее изучены. Тем не менее, недавние исследования показали, что передача сигналов IFN типа I важна для регуляции миграции миелоидных клеток во время вирусных инфекций [17,31,33], утверждая, что IFNs могут играть более широкую роль в противовирусном иммунитете, помимо их хорошо установленной присущей клетке. противовирусная активность.В этом исследовании мы сообщаем, что передача сигналов IFN типа I необходима, но не достаточна для контроля всей шкалы врожденных реакций легких на IAV. С этой целью функциональное взаимодействие между сигнальными путями IFN и IFN-γ типа I необходимо для регуляции легочного воспаления, управляемого как моноцитами, так и нейтрофилами (рис. 6F). Открытие того, что в отсутствие передачи сигналов IFN типа I IFN-γ способствует воспалительным функциям Ly6C lo Mo / Mϕ, предполагает, что взаимодействие между врожденными и адаптивными IFN определяет исход воспаления ткани для устойчивости к инфекции IAV.
Нарушенная миграция моноцитов Ly6C hi , наблюдаемая у инфицированных мышей Ifnar1 — / — , имеет сходство с мышами Ccr2 — / — [10,18,34,35]. Действительно, все лиганды CCR2 CCL2, CCL7 и CCL12 являются IFN-индуцируемыми I типа и влияют на рекрутирование моноцитов в модели хронического воспаления [36]. Ключевое различие между мышами Ccr2 — / — и Ifnar1 — / — состоит в том, что дефицит CCR2 благоприятен для исхода инфекции [10,18], тогда как Ifnar1 — / — Дефицит приводит к повышенной смертности по сравнению с животными дикого типа [17].Следовательно, IFN типа I должны опосредовать другие защитные механизмы помимо рекрутирования Ly6C hi CCR2 + воспалительных моноцитов при IAV-инфекции.
Неожиданно мы обнаружили, что передача сигналов IFN типа I играет важную роль в подавлении активации мононуклеарных клеток после инфекции IAV. Интересно, что это ингибирование особенно эффективно при подавлении провоспалительного потенциала Ly6C lo Mo / Mϕ. Наше наблюдение, что Ifnar1 — / — Ly6C lo Mo / Mϕ экспрессируют более высокие уровни CD11c, MHC класса II и NOS2, чем их аналоги Ly6C hi (WT или Ifnar1 — / — ). неожиданно.Эти клетки Ly6C lo напоминают как фенотипически, так и функционально воспалительные моноциты или дендритные клетки, продуцирующие TNF / iNOS (Tip-DC), описанные при других воспалительных состояниях (обзор в [12]). Хотя моноциты Ly6C hi и Ly6C lo считаются фенотипически разными линиями, и их созревание в процессе развития все еще вызывает споры [37,38], наши данные демонстрируют, что оба подмножества могут быть активированы IFN-γ, чтобы опосредовать пролиферацию. воспалительные действия.Принимая во внимание эти результаты, остается установить, представляют ли CD11b + Ly6C lo Mo / Mϕ в легких IAV-инфицированных мышей отдельную линию или развились из моноцитов Ly6C hi в результате подавления регуляции Экспрессия Ly6C при проникновении в воспаленные ткани легких, как описано в других моделях [33,36]. Однако последний сценарий вряд ли будет основным механизмом, объясняющим накопление Ly6C lo Mo / Mϕ в отсутствие передачи сигналов IFN типа I в нашем исследовании из-за значительной разницы в количестве общих CD11b + Mo / Mϕ в легких инфицированных мышей WT и Ifnar1 — / — .
Наш вывод о том, что IFN типа I по-разному регулирует перенос нейтрофилов и моноцитов при IAV-инфекции, согласуется с предыдущим сообщением [17]. Однако, в то время как в предыдущем исследовании анализировались исключительно клетки ЖБАЛ, в нашем исследовании изучались миелоидные популяции как в ЖБАЛ, так и в тканях легких. Кроме того, в отличие от Seo et al, мы не наблюдали каких-либо серьезных изменений в клетках-предшественниках или зрелых миелоидных клетках в BM и крови инфицированных IAV Ifnar1 — / — или смешанных химерных мышей BM, что позволяет предположить, что IFN играют играет важную роль в координации регионального иммунитета, а не центрального миелопоэза, как было предложено Seo и коллегами [17].Это несоответствие может быть объяснено тем фактом, что в предыдущем исследовании были проанализированы популяции миелоидных предшественников после инфицирования BM IAV in vitro , события, которое обычно не происходит при естественной инфекции [39].
Важно отметить, что в дополнение к определению функции IFN типа I мы обнаружили две новые функции IFN-γ в легочном ответе на инфекцию IAV; ингибирование миграции нейтрофилов и индукция активации моноцитов. Интересно, что хотя известно, что IFN-γ вызывает прямую противовирусную активность в инфицированных клетках [16] и вырабатывается в больших количествах после инфицирования гриппом, его функция при инфекции IAV остается неуловимой.Многочисленные исследования, изучающие функцию лимфоцитов, вирусный клиренс или выживаемость мышей, дефицитных по IFN-γ или IFN-γR1, сообщают об отсутствии заметных различий по сравнению с животными дикого типа [20,40–42]. Таким образом, настоящее исследование выявляет функциональную роль IFN-γ в инфекции гриппа и предполагает, что некоторые функции IFN-γ маскируются IFN-зависимыми регуляторными механизмами I типа. Роль интерферонов типа III в легочном воспалении не рассматривалась в этом отчете, возможно, что цитокины также играют роль в контроле врожденного переноса клеток и активации во время инфекции IAV.Действительно, недавнее исследование показало, что IFN типа III могут регулировать миграцию и функцию нейтрофилов в экспериментальной модели артрита [43]. Интересно, что хотя показано, что IFN типа III опосредуют иммунитет к вирусным инфекциям, существует большая степень дублирования с IFN типа I [44–46]. Будущие исследования, посвященные вовлечению IFN типа III в этот процесс, могут дать дальнейшее понимание регуляторных функций системы IFN.
Ранее сообщалось, что продукция IFN типа I подавляет иммунные ответы, вызванные IFN-γ, и устойчивость к внутриклеточным бактериям [25,26], но неизвестно, активируется ли аналогичный механизм при вирусной инфекции.В этом исследовании мы демонстрируем, что регуляторная цепь IFN также играет ключевую роль в ответе хозяина на инфекцию IAV, особенно в предотвращении Ly6C lo Mo / Mϕ от IFN-γ-индуцированной активации путем регулирования экспрессии IFN-γR1. Жестко контролируемая передача сигналов IFN-γ в Ly6C lo Mo / Mϕ может объяснить, почему при некоторых обстоятельствах эта популяция моноцитов дифференцируется в альтернативно активированные макрофаги [24], процесс, который, как известно, чувствителен к супрессии IFN-γ [47].Интересно, что описанный здесь механизм перекрестной регуляции IFN типа I, по-видимому, действует более активно в некоторых компонентах иммунного ответа на инфекцию IAV, чем в других, поскольку ингибирующий эффект IFN-γ на миграцию нейтрофилов не подавляется во время инфекции IAV. Тем не менее, это и предыдущие исследования в совокупности предполагают, что ингибирование функции IFN-γ посредством передачи сигналов IFN типа I является важным регуляторным механизмом, действующим при некоторых инфекционных и воспалительных условиях, когда продуцируются IFN как I, так и II типа [48].Мы предполагаем, что в отличие от внутриклеточной бактериальной инфекции, когда активация инфицированных моноцитов IFN-γ важна для выведения патогенов, ингибирование IFN-γ IFN типа I во время инфекции гриппа выполняет защитную роль для хозяина. Действительно, IFN-γ-индуцируемый оксид азота (NO), как было показано, играет важную роль в опосредовании легочной патологии при инфекции IAV [13,49–51] и, как таковой, должен строго контролироваться, чтобы ограничить иммуноопосредованное повреждение тканей. .
IFNs сильно индуцируются вирусными патогенами и опосредуют иммунитет хозяина к инфекциям.В этом исследовании мы демонстрируем взаимодействие и перекрестную регуляцию между сигнальными путями IFN I и II типов, координирующими многогранный воспалительный ответ легких на инфекцию IAV. Интересно, что известно, что у вирусов появились механизмы противодействия системе IFN хозяина [52]. Таким образом, наши результаты показывают, что в дополнение к нарушению внутренних противовирусных эффекторных функций клетки, как предполагалось ранее [53], блокада продукции IFN типа I или передачи сигналов вирусными продуктами может привести к нарушению регуляции воспаления, тем самым способствуя снижению устойчивости к болезням и, возможно, повышенная передача вируса.Эта гипотеза может объяснить, почему некоторые высоковирулентные штаммы IAV связаны с гипервоспалительными реакциями [54,55], и предполагает, что целенаправленное манипулирование сигнальными путями IFN может открыть новые терапевтические возможности.
Материалы и методы
Мыши
мышей C57Bl / 6 (CD45.2 + ) и CD45.1 + (B6.SJL-Ptprca) были получены из Центра ресурсов животных (ARC, Перт). Rag2 — / — , Ifnar1 — / — , Ifngr1 — / — и Stat1 — / — Bl (все background) были выведены и содержались в лаборатории Bosch Rodent Facility Сиднейского университета.
Заявление об этике
Вся работа с мышами выполнялась в соответствии с этическими принципами, установленными Комитетом по этике животных Сиднейского университета. Все эксперименты в этой рукописи были одобрены под номерами протоколов 2013/5847 и 2013/5848. Руководящие принципы Комитета по этике животных Сиднейского университета соответствуют Австралийскому кодексу ухода за животными и их использования в научных целях (2013 г.), установленного Национальным советом Австралии по здравоохранению и медицинским исследованиям.
Инфекция, вызванная вирусом гриппа A
Мышей анестезировали внутрибрюшинно (т.е.p.) инъекция 2% 2-2-трибромэтанола (авертин) и инокулирование интраназально (в) 20 бляшкообразующими единицами (БОЕ) вируса гриппа A штамма PR8 (A / Puerto Rico / 8/1934 h2N1) в объеме 50 мкл. Вирус PR8 был любезным подарком от профессора Джона Стамбаса (Университет Дикин, Виктория, Австралия).
Анализ бляшек вируса гриппа A
IAV определяли количественно с помощью анализов бляшек с клетками MDCK с использованием стандартных методов. Вкратце, 0,9 × 10 6 клеток MDCK высевали в каждую лунку 6-луночного культурального планшета.Легкие гомогенизировали в RPMI и осветляли центрифугированием в течение 5 минут при 2000 g. Гомогенаты серийно разводили в RPMI и добавляли к монослоям клеток MDCK. После инкубации в течение 45 минут при 37 ° C клетки покрывали 1% мас. / Об. Avicel (FMC Biopolymer) в среде L15 (Sigma Aldrich), содержащей 2 мкг / мл трипсина, обработанного TPCK (Worthington Biochemicals), и инкубировали при 37 ° C. , 5% СО2 в течение 3 дней. Затем клетки промывали, фиксировали метанолом и окрашивали кристаллическим фиолетовым перед подсчетом бляшек.
Изоляция ячеек
Эвтаназии животных перфузировали 10 мл PBS и легкие удаляли в 1 мл холодного 2% FCS / RPMI. Суспензии отдельных клеток получали путем диссоциации легких лезвием скальпеля и затем инкубировали в 2% FCS / RPMI с добавлением 2 мг / мл ДНКазы I (Sigma Aldrich) и коллагеназы IV (Sigma Aldrich) в течение 30 минут при 37 ° C. Затем переваренные легкие диссоциировали через сетчатый фильтр для клеток 70 мкм (Falcon) и эритроциты лизировали буфером для лизиса ACK (Life Technologies).Клетки подсчитывали с использованием исключения трипанового синего.
Что касается клеток костного мозга, бедренные и большеберцовые кости удаляли и очищали от плоти перед вымыванием костного мозга с помощью 2% FCS / RPMI. Клетки осаждали при 300 г в течение 5 минут и ресуспендировали в буфере для лизиса ACK (Life Technologies) в течение 1 минуты для лизиса эритроцитов. Клетки промывали и ресуспендировали в 2% FCS / RPMI перед подсчетом по исключению трипанового синего.
Для сбора БАЛ катетер, прикрепленный к трехходовому запорному крану, и шприц объемом 5 мл вводили в трахею, и легкие промывали 5 x 1 мл холодного PBS с добавлением 2 мМ EDTA.Клетки осаждали при 300 г в течение 5 минут, и клетки ресуспендировали в 250 мкл 2% FCS / RPMI и хранили при 4 ° C до использования для проточного цитометрического анализа.
Генерация химерных мышей костного мозга
ХимерыBM были получены путем смертельного облучения мышей-реципиентов CD45.1 + дозой 10 Гр с последующим внутривенным переносом 2 × 10 6 BM клеток от Ifnar1 — / — мышей в хвостовую часть. Для смешанных химер BM, облученных CD45.1 + реципиентных мышей WT восстановили с использованием всего 2 x 10 6 ВМ клеток из WT (CD45.1 + ) и Ifnar1 — / — (CD45.2 + ) мышей в соотношении 1: 1. Мыши получали питьевую воду с добавлением антибиотиков (триметоприм сульфата) в течение 3 недель после облучения. Мышей использовали по крайней мере через 8 недель после восстановления костного мозга.
In vivo Нейтрализация IFN-γ у химерных мышей BMДля нейтрализации in vivo IFN-γ мыши получали 500 мкг моноклонального антитела XMG1 против IFN-γ.2 внутривенно на 2 и 6 дни после инфицирования IAV. Гибридома анти-IFN-γ клона XMG1.2 была размножена в 10% FCS / RPMI с добавлением Pen / Strep (Gibco) и аффинно очищена с использованием гранул протеина G (GE Healthcare). Очищенные антитела диализовали в PBS и стерилизовали фильтрованием перед инъекцией мышам.
Проточная цитометрия
Клетки легких (1 × 10 6 ), крови (200 мкл цельной крови) или BM (4 × 10 6 ) окрашивали в соответствии со стандартными процедурами. Вкратце, клетки инкубировали в течение 30 минут с УФ-красителем Live / Dead в соответствии с инструкциями производителя (Life Technologies).Клетки окрашивали следующими антителами в отмывке FACS (2% FCS / PBS): CD4 (клон GK1.5), CD8 (53–6,7), B220 (RA3-6B2), IA / IE (M5-114.15.2). , Ly6G (1A8), Ly6C (HK1.4), CD11b (M1 / 70), CD45.1 (A20), CD45.2 (104), NK1.1 (PK136), Siglec-F (E50-2440), CCR2 (475301), LFA-1 (M17 / 4), CD119 (2E2), CD11c (N418), CD16 / 32 (93), CD64 (X54-5 / 7.1), CD45 (30-F11), коктейль Lineage, CD117 (2B8), Sca1 (D7), CD48 (HM48-1), CD150 (Q38-480). Стратегия стробирования, используемая для идентификации популяций клеток, показана на S6 Рис.Популяции моноцитов и макрофагов были идентифицированы как CD4 — CD8 — B220 — NK1.1 — SiglecF — Ly6G — CD11b + и затем классифицированы на Ly6C hi и Ly6C lo . популяции на основе экспрессии Ly6C. Нейтрофилы были идентифицированы как CD4 — CD8 — B220 — NK1.1 — SiglecF — CD11b + Ly6G + .
Для внутриклеточного окрашивания NOS2 суспензии отдельных клеток инкубировали при 37 ° C в течение 3 часов перед окрашиванием антителами к поверхностным маркерам.Внутриклеточное окрашивание проводили с использованием набора BD Cytofix / Cytoperm в соответствии с инструкциями производителя (BD). Внутриклеточное окрашивание проводили с использованием антитела NOS2 (клон CXNFT). Сбор всех данных проточной цитометрии выполнялся на LSRII с использованием программного обеспечения FACSDiva (BD Biosciences), а весь анализ проводился с использованием FlowJo X v0.7 (TreeStar).
Подготовка мРНК ткани и qRT-PCR
После перфузии ткань легких мыши собирали и погружали в RNAlater (Ambion) на 24 часа перед длительным хранением при -80 ° C.РНК получали из легких мышей с использованием Trisure (Bioline) в соответствии с инструкциями производителя (Bioline). Общую РНК (2 мкг) подвергали обратной транскрипции с использованием набора для синтеза кДНК Tetro со случайными праймерами в соответствии с инструкциями производителя (Bioline). Данные выражены в виде кратного увеличения по сравнению с неинфицированным контролем и рассчитаны методом ΔΔCT с использованием 18S в качестве контрольного гена.
Для абсолютного количественного определения вирусных нуклеопротеинов РНК экстрагировали из 1 x 10 7 PFU PR8 с использованием набора ISOLATEII RNA согласно инструкциям производителя (Bioline) и 100 нг подвергали обратной транскрипции с помощью набора для синтеза кДНК Tetro с использованием нуклеопротеин-специфичных праймеров IAV [ 39].После амплификации продукт кДНК размером 216 п.н. очищали на геле с использованием набора для экстракции геля (Sigma Aldrich) и определяли общее количество копий на основании размера и выхода продукта. Стандартная кривая была построена для определения абсолютного числа копий мРНК вирусного нуклеопротеина среди мРНК образца.
Все количественные ПЦР с обратной транскриптазой (qRT-PCR) выполняли с использованием мастер-микса SYBR NoROX (Bioline) на Roche LightCycler480. Прямые и обратные праймеры для qRT-PCR перечислены в таблице 1.
Статистика
Все статистические анализы были выполнены в Prism 6 (программное обеспечение GraphPad).Достоверность определялась с помощью теста Стьюдента t при сравнении двух экспериментальных групп или однофакторного дисперсионного анализа с последующей поправкой Тьюки для более чем двух групп. Статистически значимыми считались результаты с p <0,05. * <0,05, ** <0,01, *** <0,001, **** <0,0001.
Вспомогательная информация
S1 Рис. Экспрессия Sca1 регулируется передачей сигналов IFN.
Типичные графики проточной цитометрии, изображающие долю клеток Sca1 + в BM мышей WT и Stat1 — / — на d7 p.i .. Данные представляют 2 независимых эксперимента (n = 4–6 мышей).
https://doi.org/10.1371/journal.ppat.1005378.s001
(TIF)
S2 Рис. WT и
Ifnar1 — / — популяции мононуклеарных клеток обнаруживают сходные фенотипы сразу после инфицирования IAV.Типичные гистограммы потока, изображающие экспрессию поверхностных молекул на субпопуляциях клеток Ly6C hi и Ly6C lo в легких инфицированных d3 мышей WT и Ifnar1 — / — мышей (n = 3 мыши / генотип).Показанные данные проточной цитометрии привязаны к клеточным популяциям CD11b + CD4 — CD8 — B220 — NK1.1 — Ly6G — SiglecF — .
https://doi.org/10.1371/journal.ppat.1005378.s002
(TIF)
S3 Рис.
Ifnar1 — / — Mo / Mϕ демонстрируют повышенную экспрессию NOS2.(A) Типичные графики потока, показывающие процентное содержание NOS2 и I-A CD45.1 + (WT) и CD45.2 + ( Ifnar1 — / — ) Ly6C hi моноцитов в легких смешанных химерных мышей BM на d7 пи. Показанные данные проточной цитометрии привязаны к CD11b + CD4 — CD8 — B220 — NK1.1 — Ly6G — SiglecF — популяции клеток. Смешанные химерные мыши BM были созданы, инфицированы и проанализированы, как описано на фиг. 2D. Данные представляют 2 независимых эксперимента (n = 5). (B) Парный анализ средней интенсивности флуоресценции (MFI) NOS2 в популяциях Ly6C hi и Ly6C lo инфицированных смешанных мышей-химер BM на d7 p.i .. Данные были объединены из 2 независимых экспериментов. Статистический анализ проводили с использованием парного теста Стьюдента t .
https://doi.org/10.1371/journal.ppat.1005378.s003
(TIF)
S4 Рис. Процент нейтрофилов Ly6G
+ в крови наивных смешанных мышей-химер BM.Смешанные CD45.1 + (WT) и CD45.2 + ( Ifnar1 — / — ) BM химерных мышей были созданы, инфицированы и проанализированы с использованием проточной цитометрии, как описано на фиг. 2D.Данные показывают процент нейтрофилов CD45.1 + (WT) и CD45.2 + ( Ifnar1 — / — ) Ly6G + нейтрофилов в крови наивных смешанных химерных мышей BM. Данные являются репрезентативными для 2 независимых экспериментов (n = 10–14 мышей / исследование).
https://doi.org/10.1371/journal.ppat.1005378.s004
(TIF)
S5 Рис. Нарушение миграции моноцитов в инфицированных IAV
Ifnar1 — / — химерных мышах.Сводные данные, отражающие процентное содержание мононуклеарных клеток Ly6C hi и Ly6C lo в легких IAV-инфицированных WT, Ifnar1 — / — и CD45.1 + WT / CD45.2 + Ifnar1 — / — химерных мышей d7 pi. Восстановленные химерные мыши Ifnar1 — / — BM были получены, инфицированы и проанализированы, как описано на рис. Данные представляют 2 независимых эксперимента.
https://doi.org/10.1371/journal.ppat.1005378.s005
(TIF)
S6 Рис. Стратегия стробирования, используемая для идентификации популяций клеток легких.
Типичные графики проточной цитометрии суспензий единичных клеток легких с указанием стробирования, используемого для идентификации основных популяций лейкоцитов легких.Показанные графики потока были получены с 7-го дня в день. Мышь WT.
https://doi.org/10.1371/journal.ppat.1005378.s006
(TIF)
Благодарности
Мы благодарим Анхеля Панга за техническую помощь, A / Prof. Джон Стамбас за предоставление IAV и докторов. Эндрю Митчеллу и Фредерику Сьерро за критическое прочтение рукописи.
Вклад авторов
Задумал и спроектировал эксперименты: SAS CGF. Проведены эксперименты: САС НБ РП МФ.Проанализированы данные: SAS JAT WJB CGF. Предоставленные реагенты / материалы / инструменты анализа: MF JAT WJB CGF. Написал статью: SAS JAT WJB CGF.
Ссылки
- 1. Braciale TJ, Sun J, Kim TS (2012) Регулирование адаптивного иммунного ответа на респираторную вирусную инфекцию. Нат Рев Иммунол 12: 295–305. pmid: 22402670
- 2. Ивасаки А., Пиллаи П.С. (2014) Врожденный иммунитет к инфекции вируса гриппа. Nat Rev Immunol 14: 315–328. pmid: 24762827
- 3.Кобаса Д., Джонс С.М., Шинья К., Каш Дж. К., Коппс Дж. И др. (2007) Аберрантный врожденный иммунный ответ при летальном инфицировании макак вирусом гриппа 1918 г. Природа 445: 319–323. pmid: 17230189
- 4. Каш Дж. К., Баслер К.Ф., Гарсия-Састре А., Картер В., Биллхарц Р. и др. (2004) Глобальный иммунный ответ хозяина: патогенез и транскрипционное профилирование вирусов гриппа типа А, экспрессирующих гены гемагглютинина и нейраминидазы из пандемического вируса 1918 года. J Virol 78: 9499–9511.pmid: 15308742
- 5. Kash JC, Tumpey TM, Proll SC, Carter V, Perwitasari O и др. (2006) Геномный анализ повышенной иммунной реакции хозяина и реакции гибели клеток, вызванной вирусом гриппа 1918 года. Природа 443: 578–581. pmid: 17006449
- 6. La Gruta NL, Kedzierska K, Stambas J, Doherty PC (2007) Вопрос самосохранения: иммунопатология при инфекции вируса гриппа. Immunol Cell Biol 85: 85–92. pmid: 17213831
- 7. Brandes M, Klauschen F, Kuchen S, Germain RN (2013) Системный анализ выявляет прямую воспалительную цепь, ведущую к летальной инфекции гриппа.Cell 154: 197–212. pmid: 23827683
- 8. Нарасараджу Т., Ян Э., Сами Р.П., Нг Х.Х., Пох В.П. и др. (2011) Избыточные нейтрофилы и внеклеточные ловушки нейтрофилов способствуют острому повреждению легких при гриппозном пневмоните. Am J Pathol 179: 199–210. pmid: 21703402
- 9. Narasaraju T, Ng HH, Phoon MC, Chow VT (2010) Лечение антителом MCP-1 усиливает повреждение и препятствует восстановлению альвеолярного эпителия при гриппозном пневмоните. Am J Respir Cell Mol Biol 42: 732–743.pmid: 19617401
- 10. Lin KL, Suzuki Y, Nakano H, Ramsburg E, Gunn MD (2008) Дендритные клетки, полученные из моноцитов CCR2 +, и макрофаги экссудата вызывают индуцированную гриппом легочную иммунную патологию и смертность. J Immunol 180: 2562–2572. pmid: 18250467
- 11. Гейссманн Ф., Манц М.Г., Юнг С., Сивеке М.Х., Мерад М. и др. (2010) Развитие моноцитов, макрофагов и дендритных клеток. Наука 327: 656–661. pmid: 20133564
- 12. Shi C, Pamer EG (2011) Рекрутирование моноцитов во время инфекции и воспаления.Nat Rev Immunol 11: 762–774. pmid: 21984070
- 13. Олдридж-младший, Мозли К.Э., Больц Д.А., Неговетич Н.Дж., Рейнольдс С. и др. (2009) Дендритные клетки, продуцирующие TNF / iNOS, являются неизбежным злом смертельной инфекции вируса гриппа. Proc Natl Acad Sci U S A 106: 5306–5311. pmid: 1 09
- 14. Auffray C, Sieweke MH, Geissmann F (2009) Моноциты крови: развитие, гетерогенность и взаимосвязь с дендритными клетками. Анну Рев Иммунол 27: 669–692. pmid: 117
- 15.Пестка С. (2007) Интерфероны: через 50 лет после их открытия предстоит еще многое узнать. J Biol Chem 282: 20047–20051. pmid: 17502369
- 16. Schroder K, Hertzog PJ, Ravasi T, Hume DA (2004) Интерферон-гамма: обзор сигналов, механизмов и функций. Журнал биологии лейкоцитов 75: 163–189. pmid: 14525967
- 17. Сео СУ, Квон Х.Дж., Ко Х.Дж., Бюн Й.Х., Сеонг Б.Л. и др. (2011) Передача сигналов интерферона типа I регулирует моноциты и нейтрофилы Ly6C (hi) во время острой вирусной пневмонии у мышей.PLoS Pathog 7: e1001304. pmid: 21383977
- 18. Lin SJ, Lo M, Kuo RL, Shih SR, Ojcius DM, et al. (2014) Патологические эффекты воспалительных моноцитов CCR2 + усиливаются петлей обратной связи хемокинов, запускаемой IFNAR1, при высокопатогенной инфекции гриппа. J Biomed Sci 21: 99. pmid: 25407417
- 19. Marois I, Cloutier A, Garneau E, Richter MV (2012) Начальная инфекционная доза определяет врожденную, адаптивную реакцию и реакцию памяти на грипп в дыхательных путях.J Leukoc Biol 92: 107–121. pmid: 22504848
- 20. Гарсия-Састре А., Дурбин Р.К., Чжэн Х., Палезе П., Гертнер Р. и др. (1998) Роль интерферона в тропизме тканей вируса гриппа. J Virol 72: 8550–8558. pmid: 9765393
- 21. Дурбин Дж. Э., Фернандес-Сесма А., Ли С. К., Рао Т. Д., Фрей А. Б. и др. (2000) IFN типа I модулирует врожденный и специфический противовирусный иммунитет. J Immunol 164: 4220–4228. pmid: 10754318
- 22. Essers MAG, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, et al.(2009) IFN-альфа активирует спящие гемопоэтические стволовые клетки in vivo. Nature 458: 904 – U911. pmid: 121
- 23. Oguro H, Ding L, Morrison SJ (2013) Маркеры семейства SLAM разрешают функционально разные субпопуляции гемопоэтических стволовых клеток и мультипотентных предшественников. Стволовые клетки клетки 13: 102–116. pmid: 23827712
- 24. Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O и др. (2007) Мониторинг кровеносных сосудов и тканей популяцией моноцитов с патрулирующим поведением.Наука 317: 666–670. pmid: 17673663
- 25. Антонелли Л. Р., Джильотти Ротфукс А., Гонсалвес Р., Роффе Е., Чивер А. В. и др. (2010) Интраназальное лечение Poly-IC обостряет туберкулез у мышей из-за рекрутирования в легкие популяции моноцитов / макрофагов, допускающих патогенез. Дж. Клин Инвест 120: 1674–1682. pmid: 20389020
- 26. Rayamajhi M, Humann J, Penheiter K, Andreasen K, Lenz LL (2010) Индукция IFN-алфавита позволяет Listeria monocytogenes подавлять активацию макрофагов IFN-гамма.J Exp Med 207: 327–337. pmid: 20123961
- 27.
Кирни С.Дж., Дельгадо С., Эшлеман Э.М., Хилл К.К., О’Коннор Б.П. и др. (2013) IFN типа I подавляют рецептор IFN-гамма миелоидных клеток, вызывая рекрутирование комплекса 3 / NGFI-A-связывающий белок 1, который подавляет транскрипцию ifngr1. J Immunol 191: 3384–3392. pmid: 23
7 - 28. Silvennoinen O, Schindler C, Schlessinger J, Levy DE (1993) Ras-независимая передача сигналов фактора роста посредством фосфорилирования тирозина транскрипционного фактора.Наука 261: 1736–1739. pmid: 8378775
- 29. Lee CK, Gimeno R, Levy DE (1999) Дифференциальная регуляция экспрессии конститутивного главного комплекса гистосовместимости класса I в T- и B-лимфоцитах. J Exp Med 190: 1451–1464. pmid: 10562320
- 30. Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L (1998) Передача сигналов цитокина типа интерлейкина-6 через путь gp130 / Jak / STAT. Biochem J 334 (Pt 2): 297–314. pmid: 9716487
- 31. Stock AT, Smith JM, Carbone FR (2014) IFN типа I подавляет рекрутирование нейтрофилов, вызванное Cxcr2, в сенсорные ганглии во время вирусной инфекции.J Exp Med 211: 751–759. pmid: 24752295
- 32. Samuel CE (2001) Противовирусное действие интерферонов. Clin Microbiol Rev 14: 778–809, содержание. pmid: 11585785
- 33. Горицка М., Макрис С., Каусар Ф., Дюрант Л. Р., Перейра С. и др. (2015) Интерфероны I типа, происходящие из альвеолярных макрофагов, организуют врожденный иммунитет к RSV за счет привлечения противовирусных моноцитов. J Exp Med 212: 699–714. pmid: 25897172
- 34. Wareing MD, Lyon A, Inglis C, Giannoni F, Charo I, et al.(2007) Хемокиновая регуляция воспалительного ответа на низкую дозу инфекции гриппа у мышей CCR2 — / -. J Leukoc Biol 81: 793–801. pmid: 17179466
- 35. Dawson TC, Beck MA, Kuziel WA, Henderson F, Maeda N (2000) Контрастные эффекты дефицита CCR5 и CCR2 в воспалительной реакции легких на вирус гриппа А. Am J Pathol 156: 1951–1959. pmid: 10854218
- 36. Ли ПЙ, Ли Й, Кумагаи И, Сюй И, Вайнштейн Дж. С. и др. (2009) Интерферон типа I модулирует рекрутирование и созревание моноцитов при хроническом воспалении.Am J Pathol 175: 2023–2033. pmid: 19808647
- 37. Ханна Р.Н., Карлин Л.М., Хаббелинг Х.Г., Нацкевич Д., Грин А.М. и др. (2011) Фактор транскрипции NR4A1 (Nur77) контролирует дифференцировку костного мозга и выживание моноцитов Ly6C-. Nat Immunol 12: 778–785. pmid: 21725321
- 38. Йона С., Ким К.В., Вольф Ю., Милднер А., Варол Д. и др. (2013) Картирование судьбы показывает происхождение и динамику моноцитов и тканевых макрофагов в условиях гомеостаза. Иммунитет 38: 79–91.pmid: 23273845
- 39. Hermesh T, Moltedo B, Moran TM, Lopez CB (2010) Противовирусная инструкция лейкоцитов костного мозга во время респираторных вирусных инфекций. Клеточный микроб-хозяин 7: 343–353. pmid: 20478536
- 40. Грэм МБ, Далтон Д.К., Гилтинан Д., Брасиале В.Л., Стюарт Т.А. и др. (1993) Ответ на инфекцию гриппа у мышей с направленным нарушением гена интерферона гамма. J Exp Med 178: 1725–1732. pmid: 8228818
- 41. Price GE, Gaszewska-Mastarlarz A, Moskophidis D (2000) Роль альфа / бета- и гамма-интерферонов в развитии иммунитета к вирусу гриппа A у мышей.J Virol 74: 3996–4003. pmid: 10756011
- 42. Нгуен Х.Х., ван Гинкель Ф.В., Ву Х.Л., Новак М.Дж., МакГи Дж.Р. и др. (2000) Гамма-интерферон не требуется для цитотоксических Т-лимфоцитов слизистой оболочки или гетеросубтипического иммунитета к инфекции вируса гриппа А у мышей. J Virol 74: 5495–5501. pmid: 10823854
- 43. Блазек К., Имс Х.Л., Вайс М., Бирн А.Дж., Перошо Д. и др. (2015) IFN-lambda устраняет воспаление за счет подавления инфильтрации нейтрофилов и выработки IL-1beta.J Exp Med 212: 845–853. pmid: 255
- 44. Mordstein M, Kochs G, Dumoutier L, Renauld JC, Paludan SR, et al. (2008) Интерферон-лямбда способствует врожденному иммунитету мышей против вируса гриппа A, но не против гепатотропных вирусов. PLoS Pathog 4: e1000151. pmid: 18787692
- 45. Wack A, Terczynska-Dyla E, Hartmann R (2015) На страже границ: биология интерферонов типа III. Нат Иммунол 16: 802–809. pmid: 261
- 46. Lazear HM, Nice TJ, Diamond MS (2015) Интерферон-лямбда: иммунные функции на барьерных поверхностях и за их пределами.Иммунитет 43: 15–28. pmid: 26200010
- 47. Гордон С., Мартинес Ф.О. (2010) Альтернативная активация макрофагов: механизм и функции. Иммунитет 32: 593–604. pmid: 20510870
- 48. Stifter SA, Feng CG (2015) Вмешательство в иммунитет: пагубная роль IFN типа I во время инфекции. J Immunol 194: 2455–2465. pmid: 25747907
- 49. Karupiah G, Chen JH, Mahalingam S, Nathan CF, MacMicking JD (1998) Быстрый интерферон-гамма-зависимый клиренс вируса гриппа A и защита от консолидации пневмонита у мышей с дефицитом синтазы оксида азота 2.J Exp Med 188: 1541–1546. pmid: 9782132
- 50. Акаике Т., Ногучи Ю., Иджири С., Сетогучи К., Суга М. и др. (1996) Патогенез пневмонии, вызванной вирусом гриппа: участие как оксида азота, так и кислородных радикалов. Proc Natl Acad Sci U S A 93: 2448–2453. pmid: 8637894
- 51. Perrone LA, Belser JA, Wadford DA, Katz JM, Tumpey TM (2013) Индуцируемый оксид азота способствует вирусному патогенезу после инфицирования мышей высокопатогенным вирусом гриппа.J Infect Dis 207: 1576–1584. pmid: 23420903
- 52. Гарсия-Састре А., Бирон CA (2006) Интерфероны типа 1 и отношения вирус-хозяин: урок разрядки. Наука 312: 879–882. pmid: 166
- 53. Kochs G, Garcia-Sastre A, Martinez-Sobrido L (2007) Множественные антиинтерфероновые действия белка NS1 вируса гриппа А. J Virol 81: 7011–7021. pmid: 17442719
- 54. Perrone LA, Plowden JK, Garcia-Sastre A, Katz JM, Tumpey TM (2008) Инфекция вируса пандемического гриппа H5N1 и 1918 года приводит к ранней и чрезмерной инфильтрации макрофагов и нейтрофилов в легкие мышей.PLoS Pathog 4: e1000115. pmid: 18670648
- 55. Cilloniz C, Shinya K, Peng X, Korth MJ, Proll SC и др. (2009) Смертельная инфекция вирусом гриппа у макак связана с ранним нарушением регуляции генов, связанных с воспалением. PLoS Pathog 5: e1000604. pmid: 19798428
Противовирусная и иммунорегуляторная активность IFN-γ зависит от конститутивно экспрессируемого IL-1α
Аннотация
IFN-γ индуцирует свою иммунорегуляторную активность, активируя гены, главным образом, через сигнальный путь Jak-STAT.Здесь мы показываем, что то, что считалось внутренней активностью IFN-γ, в значительной степени зависит от базального уровня NF-κB, который поддерживается конститутивно экспрессируемым IL-1α. Антагонист рецептора IL-1 и антитела к IL-1α, но не к IL-1β, ингибировали противовирусную активность IFN-γ на 90%, тогда как ингибирования активности IFN типа I не наблюдалось. Точно так же индукция многих генов IFN-γ, включая HLA-DR , ICAM-1 , IL-18BP и гены, опосредующие его противовирусную активность, в значительной степени зависела от базального IL-1α.Кроме того, IFN-γ индуцировал сывороточный белок, связывающий IL-18, у мышей дикого типа, но не у мышей с двойным дефицитом IL-1α / β. Таким образом, конститутивно экспрессируемый IL-1α имеет решающее значение для многих активностей IFN-γ.
Первоначально идентифицированный по своей противовирусной активности в неиммунных клетках (1), IFN-γ, как было позже обнаружено, регулирует значительный диапазон иммунных ответов и является главным регулятором ответа Th2 (2–4). Большинство типов клеток экспрессируют рецептор IFN-γ (5), и его связывание активирует STAT1, который перемещается в виде димера в ядро, активируя гены, промотор которых содержит γ-активированную последовательность (GAS) (6).Некоторые из этих IFN-γ-индуцированных генов являются регуляторами транскрипции, такими как IFN-регуляторный фактор (IRF) -1, который индуцирует другие гены, в том числе те, которые опосредуют противовирусное действие IFN-γ (7).
TNF и IL-1 усиливают транскрипционную активность IFN-γ. Сообщалось об аддитивной или даже синергической индукции нескольких провоспалительных генов, включая ICAM-1 , iNOS , различные гены хемокинов и фактор роста гепатоцитов (8-15). Аддитивная и синергическая индукция генов IFN-γ и IL-1 / TNF в значительной степени объясняется независимой активацией ответных элементов GAS и κB соответственно.Эти два элемента ответа противопоставлены во многих промоторах вышеупомянутых генов (16).
Роль базального NF-κB в действии IFN-γ изучена в ограниченной степени. Индукция генов хемокинов CXCL9 и CXCL10 с помощью IFN-γ зависит от конститутивной активации NF-κB, однако триггер этой базовой активации не был идентифицирован (17). Поскольку большинство типов клеток экспрессируют конститутивно низкие уровни IL-1α (18), мы изучили здесь его роль в индукции генов IFN-γ.
Результаты
ИнгибиторыIL-1α снижают противовирусную активность IFN-γ.
Сначала мы изучили роль эндогенного IL-1α в противовирусной активности различных IFN. Эпителиальные клетки WISH человека инкубировали с различными IFN в присутствии или в отсутствие ингибиторов IL-1. Затем культуры заражали вирусом везикулярного стоматита и оценивали способность IFN-γ защищать от гибели клеток. Когда к клеткам добавляли IFN-γ (серийно разведенный в 2 раза), в лунке 7 было 50% защиты, что соответствовало титру 4000 единиц / мл (рис.1 A , столбец 2). Добавление антагониста рецептора IL-1 (IL-1Ra) или антител против IL-1α вместе с IFN-γ снижало его противовирусный титр на 90% до 400 единиц / мл (рис. A , столбцы 3 и 4 соответственно). Напротив, антитела против IL-1β не влияли на титр противовирусного IFN-γ (столбец 5). Экзогенный IL-1α (данные не показаны) или IL-1β увеличивали противовирусную активность IFN-γ почти в 2 раза, а IL-1Ra блокировал противовирусную активность, усиленную как экзогенным, так и эндогенным IL-1α (рис.1 A , столбцы 6 и 7). IL-1α (данные не показаны) и IL-1β (столбец 8) не проявляли противовирусной активности в отсутствие IFN-γ. Напротив, ингибиторы IL-1 не влияли на противовирусную активность IFN типа I. (Рисунок 1 В ). Тем не менее, 2 нг / мл IL-1β увеличивали противовирусную активность как IFN-α2, так и IFN-β в ≈2 раза, подтверждая предыдущее исследование (данные не показаны) (19). Эти результаты показывают, что эндогенный IL-1α имеет решающее значение для противовирусной активности IFN-γ, но не для IFN типа I.
Рисунок 1.Влияние ингибиторов ИЛ-1 на противовирусную активность ИФН. Противовирусный титр различных IFN человека I и II типа определяли с помощью анализа ингибирования цитопатического эффекта вируса (вирус везикулярного стоматита). ( A ) В столбце 1 показаны контрольные клетки WISH (четыре нижние лунки) и полный цитопатический эффект тех же клеток после заражения вирусом везикулярного стоматита (четыре верхние лунки).Эпителиальные клетки WISH инкубировали с серийным 2-кратным разведением 4000 единиц / мл IFN-γ, либо отдельно, либо в присутствии экзогенных 2 нг / мл ингибиторов IL-1β или IL-1 (10 мкг / мл IL-1Ra, 2 мкг / мл антител против IL-1α или 2 мкг / мл антител против IL-1β). Титр относительно одного IFN-γ (столбец 2) дан в процентах ниже лунок. ( B ) IFN-α2 (4000 единиц / мл) или IFN-β (8000 единиц / мл) добавляли к клеткам либо отдельно, либо вместе с IL-1Ra, и титр противовирусного препарата определяли, как в A .
Чтобы дополнительно подтвердить критическую роль эндогенного IL-1 в противовирусной активности IFN-γ, эту активность оценивали в мышиных эмбриональных фибробластах (MEF) дикого типа и в MEF с двойным нокаутом IL-1α / β. Подобно клеткам WISH, IFN-γ был только 10% активен в MEF от одной мыши с двойным нокаутом по сравнению с MEF C / 57BL дикого типа и 33% активен в MEF от другой мыши с двойным нокаутом.
Анализ IFN-γ-индуцированных генов с помощью экспрессионных массивов.
Поскольку антивирусное состояние индуцируется на уровне транскрипции, мы выполнили анализ массива экспрессии, чтобы получить представление о роли IL-1α в транскрипционной активности, индуцированной IFN-γ. Клетки WISH обрабатывали IFN-γ, IL-1Ra или их комбинацией в течение разного времени, и общую РНК подвергали анализу массива экспрессии. Влияние IL-1Ra на экспрессию гена IFN-γ было минимальным через 3 часа, увеличивалось через 6 часов и было максимальным через 17 часов. Из 370 генов, индуцированных по крайней мере в 4 раза за 17 ч, мы обнаружили группу из 65 генов, индукция которых ингибировалась по крайней мере на одну треть в присутствии IL-1Ra.Среди этих генов были некоторые из основных IFN-γ-индуцированных транскриптов, связанных с противовирусным ответом, в том числе Mx1 , Mx2 , 2′-5′-олигоА-синтетаза , ISG20 и GBP1 (рис. 2) (20–23). Дополнительные известные IFN-γ-индуцированные гены, не связанные с противовирусной активностью IFN-γ, также оказались зависимыми от IL-1, включая диубиквитин ( UBD / FAT10 ), ICAM1 и MHCII. (рис.2) (3, 24, 25). Однако другие известные IFN-γ-индуцированные гены не модулируются IL-1Ra, включая компоненты комплемента C4A и C4B , рецептор IL-15 α цепь , IFI 16b , IFI 41 , и триптофанилсинтетаза (данные не показаны). Полуколичественная ОТ-ПЦР РНК из обработанных IFN-γ клеток WISH (фиг. 3A), а также кератиноцитов HaCaT (фиг. 3 B ) подтвердили данные массива экспрессии для многих из этих транскриптов, включая IRF-1 , CIITA , HLA-DR , ICAM1 , CXCL11 , IL-32 ( NK4 ) (26) и FAT10 ( UBD ).Добавление экзогенного IL-1β обращало ингибирование IL-1Ra (рис. 3). A , полосы 6 и 12). Взятые вместе, эти результаты устанавливают существование группы генов, индукция которых IFN-γ в значительной степени зависит от эндогенно экспрессируемого IL-1α.
Рис. 2.Анализ массива экспрессии IFN-γ-обработанных человеческих клеток WISH.Клетки WISH человека обрабатывали либо средой, 100 ед. / Мл IFN-γ, 10 мкг / мл IL-1Ra, либо их комбинацией. Полную РНК выделяли через 17 часов и подвергали анализу экспрессионной матрицы (чип Affymetrix U133A). С помощью программного обеспечения Excel файлы данных Affymetrix были отсортированы по алгоритму, описанному в Материалы и методы . Показана кратность индукции генов в клетках, обработанных IFN-γ в течение 17 ч в отсутствие (заштрихованные столбцы) или в присутствии (белые столбцы) IL-1Ra. Значения в скобках представляют собой средний сигнал двух повторных анализов.Программа Affymetrix считала все сигналы и кратные индукции значимыми ( P <0,05). Индукция генов и анализ массивов были выполнены дважды с очень похожими результатами.
Рис. 3.ОТ-ПЦР отобранных генов после индукции IFN-γ. Клетки WISH человека ( A ) или кератиноциты HaCaT ( B ) обрабатывали 100 ед. / Мл IFN-γ, 2 нг / мл IL-1β, 10 мкг / мл IL-1Ra или их комбинацией.РНК выделяли в указанные сроки и подвергали полуколичественной ОТ-ПЦР со специфическими праймерами. Количество циклов было скорректировано, чтобы показать максимальные различия, если таковые имеются. ОТ-ПЦР β-актина использовали для контроля загрузки образца.
IL-1Ra ингибирует индукцию IL-18-связывающего белка (IL-18BP) под действием IFN-γ.
Установив роль эндогенного IL-1α в модулировании экспрессии IFN-γ-индуцированных транскриптов, мы исследовали его действие на уровне белка.IL-18BP — это индуцированный IFN-γ ген, экспрессия которого значительно повышается при различных воспалительных заболеваниях (27). Его базальная экспрессия очень низкая в негематопоэтических клетках, и до сих пор только IFN-γ был идентифицирован как индуктор IL-18BP (28, 29). Неожиданно анализ массива экспрессии не обнаружил индукции мРНК IL-18BP IFN-γ. Однако ОТ-ПЦР подтвердила устойчивую индукцию мРНК IL-18BP IFN-γ в клетках WISH и в кератиноцитах HaCaT. С помощью ОТ-ПЦР мы обнаружили, что индукция IL-18BP в клетках WISH в значительной степени зависит от эндогенного IL-1 (рис.3). На уровне белка контрольные культуры кератиноцитов HaCaT содержали 0,39 ± 0,03 нг / мл IL-18BP, как определено с помощью ELISA. После индукции IFN-γ уровень IL-18BP повысился до 2,77 ± 0,024 нг / мл ( P = 0,0003, n = 9), тогда как индукция IFN-γ не была получена в присутствии IL- 1Ra (0,35 ± 0,003 нг / мл).
Затем мы определили роль IL-1α в индукции IL-18BP in vivo путем сравнения сывороточного IL-18BP у мышей с двойным дефицитом IL-1α / β и мышей C57BL / 6 дикого типа.Хотя обе группы мышей имели одинаковый базальный уровень циркулирующего IL-18BP, значительная индукция IL-18BP была получена после введения IFN-γ только у мышей дикого типа ( P = 0,0004, n = 8) ( Рис.4). Взятые вместе, эти результаты показывают, что эндогенный IL-1α необходим для индукции IL-18BP с помощью IFN-γ, как определено на уровнях мРНК и белка in vitro и in vivo .
Инжир.4.Сывороточный IL-18BP у мышей дикого типа и мышей с двойным нокаутом IL-1α / β после введения IFN-γ. Мышам C57BL / 6 с двойным дефицитом IL-1α / β ( n = 8 на группу) вводили внутрибрюшинно. с мышиным 50 000 единиц IFN-γ на мышь. Уровень IL-18BP определяли до введения IFN-γ и через 24 часа после введения.
Роль NF-κB в активации генов, индуцированной IFN-γ.
IFN-γ передает сигнал через путь Jak-STAT и не активирует напрямую NF-κB.Мы предположили, что эндогенный IL-1α имеет решающее значение для действия IFN-γ, обеспечивая базовый уровень активности NF-κB. Действительно, пирролидиндиокарбамат аммония (PDTC), специфический ингибитор транслокации NF-κB в ядро, полностью аннулировал индукцию мРНК IL-18BP под действием IFN-γ (рис. 5). А ). Кроме того, EMSA с зондом, соответствующим сайту κB промотора IL-18BP [основания от -396 до -387 относительно сайта начала транскрипции (28)], выявила образование комплекса с ядерными экстрактами WISH, обработанных IL-1α. клетки, которые были уничтожены IL-1Ra (рис.5 B , полосы 1–3). Этот комплекс содержал NF-κB, как показывает суперсифтинг с антителами против p65 (рис. 5). Б , пер.7).
Рис. 5.Роль IL-1α-индуцированного NF-κB в активности IFN-γ. ( A ) Клетки WISH человека обрабатывали PDTC аммония при 300 мкМ в течение 30 минут, а затем 100 ед / мл IFN-γ в течение 17 часов.Тотальную РНК выделяли и подвергали полуколичественной RT-PCR с праймерами, специфичными для человеческого IL-18BP. ( B ) Клетки WISH человека обрабатывали в течение 30 мин указанными комбинациями PDTC (300 мкМ), IL-1α (2 нг / мл), IL-1Ra (10 мкг / мл) и TNF-α (10 мкг / мл). нг / мл). Ядерные экстракты подвергали EMSA с зондом, соответствующим участку κB промотора IL-18BP. Суперсифтинг проводили с использованием антител против NF-κB p65 (дорожки 7 и 8). ( C ) К клеткам WISH добавляли серийный 2-кратно разведенный IFN-γ (100 единиц / мл), смешанный либо с 10 нг / мл TNF-α, либо без 10 мкг / мл IL-1Ra.Защиту от вирусного цитопатического эффекта оценивали, как показано на рис. 1.
Для дальнейшего установления роли NF-κB как коактиватора передачи сигналов IFN-γ мы использовали экзогенный TNF-α в качестве альтернативного индуктора NF-κB. Как и ожидалось, ядерные экстракты клеток WISH, обработанных 10 нг / мл TNF-α в течение 30 часов, также образовывали комплекс с зондом κB, который ингибировался PDTC, но не IL-1Ra (рис. 5). B , полосы 4–6). Этот TNF-α-индуцированный комплекс был идентифицирован как NF-κB путем суперсифтинга с помощью антител против p65 (рис.5 Б , переулок 8). Затем мы проверили, может ли экзогенно добавленный TNF-α заменить эндогенный IL-1α в установлении антивирусного состояния с помощью IFN-γ. Действительно, TNF-α (10 нг / мл), который не обладал внутренней противовирусной активностью, усиливал противовирусную активность IFN-γ в 32 раза (рис. 5). C ), результат соответствует предыдущим отчетам (30). Индуцированное TNF увеличение титра противовирусного препарата не ингибировалось IL-1Ra (рис. 5). C , столбец 4), демонстрируя, что TNF-α оказывает усиливающее действие на IFN-γ независимо от IL-1.Взятые вместе, эти данные предполагают, что IL-1α-активированный NF-κB играет критическую роль в экспрессии набора IFN-γ-индуцированных генов, включая те, которые опосредуют его противовирусное действие.
Интегральная мембранаIL-1α играет роль в активации генов IFN-γ.
Сообщалось, чтоIFN-γ индуцирует связанный с мембраной IL-1α в различных клетках (31, 32). Мы обнаружили, что базальный уровень связанного с клетками IL-1α составлял 10,5 ± 1,6 и 2,9 ± 0,4 пг / мкг белка в клетках WISH и кератиноцитах HaCaT, соответственно.Через 24 часа IFN-γ индуцировал IL-1α в этих клетках до 30,8 ± 6,5 и 34,9 ± 6,3 пг / мкг соответственно ( P = 0,004 и 0,001, соответственно; n = 9). Напротив, уровень IL-1α в культуральных супернатантах клеток WISH и кератиноцитов HaCaT до или после 24 часов обработки IFN-γ был ниже предела обнаружения (≈2 пг / мл).
Поскольку большая часть базального и IFN-γ-индуцированного IL-1α была клеточно-ассоциированной, мы использовали эксперименты по сокультивированию, чтобы определить, был ли он активен как интегральный мембранный белок.IL-1α индуцировался в человеческих макрофагоподобных THP-1 (не прикрепленных) клетках обработкой IFN-γ в течение 1-17 часов. Клетки промывали, фиксировали 1% параформальдегидом в течение 4 ч, снова промывали и инкубировали в течение 24 ч при 37 ° C в питательной среде. Было показано, что эта процедура индуцирует связанный с мембраной IL-1α и предотвращает утечку биологически активного про-IL-1α из внутриклеточных пулов (33). Промытые клетки THP-1 совмещали в течение 6 часов с клетками WISH в присутствии или в отсутствие IL-1Ra. После удаления клеток THP-1 степень активации NF-κB в клетках WISH оценивали с помощью EMSA с помощью γ- 32 P-меченного κB зонда.Базальная активация NF-κB наблюдалась в клетках WISH, которые были совместно культивированы с необработанными клетками THP-1, и в значительной степени индуцировалась, когда клетки WISH совпадали с клетками THP-1, предварительно обработанными IFN-γ в течение 1-17 часов (17 h, рис. 6, сравните дорожки 1 и 2). Образование комплексов, содержащих NF-κB p65, снижалось, когда сокультивирование проводилось в присутствии IL-1Ra (фиг. 6, сравните дорожки 1 и 3), тем самым идентифицируя интегральный мембранный IL-1α клеток THP-1 как важный индуктор NF-κB в клетках WISH при совместном культивировании.Такие же результаты наблюдались при совместном культивировании клеток THP-1 с клетками WISH в течение 24 часов вместо 6 часов (данные не показаны). Практически идентичные результаты наблюдались при сокультивировании клеток THP-1 с кератиноцитами HaCaT вместо клеток WISH (рис. 6, дорожки 10–12). Важно отметить, что эти данные демонстрируют, что базальная и IFN-γ-индуцированная интегральная мембрана IL-1α может действовать на соседние клетки, вызывая биологические активности юкстакринным образом.
Инжир.6.IFN-γ индуцирует активный интегральный мембранный IL-1α. Человеческие макрофагоподобные клетки THP1 либо не обрабатывали (C), либо предварительно обрабатывали 100 ед. / Мл IFN-γ в течение 17 часов (+) для индукции мембранно-ассоциированного IL-1α. Затем клетки фиксировали, как описано (33), и сокультивировали в течение 6 ч либо с клетками WISH (дорожки 1–4), либо с кератиноцитами HaCaT (дорожки 10–12) в среде отдельно или вместе с 10 мкг / мл IL-1Ra. Другие культуры WISH и HaCaT (дорожки 5–8 и 13–15 соответственно) обрабатывали указанными комбинациями IL-1α, IL-1Ra и IFN-γ.После совмещения клетки THP-1 удаляли, клетки WISH и HaCaT промывали, а их экстракты подвергали EMSA с использованием зонда κB. Контрольный экстракт фиксированных клеток THP-1 также подвергали EMSA (дорожка 9). Полосы, отмеченные стрелкой, представляют ядерные белки, специфически связывающиеся с зондом κB. Открытая стрелка указывает на более быстрый мигрирующий комплекс, который в основном содержит NF-κB p50, а закрытая стрелка указывает на более медленный мигрирующий комплекс, который содержит как NF-κB p50, так и p65.Антитело против NF-κB (p65) использовали для суперсмещения (стрелка, дорожки 4 и 15).
IL-1 ускоряет и увеличивает индукцию IRF-1 IFN-γ.
Чтобы получить представление о механизме, с помощью которого эндогенный IL-1α увеличивает активность IFN-γ, мы проверили, модулирует ли IL-1α сигнальный путь Jak-STAT. Клетки WISH инкубировали в течение 15 минут или 4 часов с IFN-γ или IL-1α, отдельно или в комбинации, в присутствии или в отсутствие IL-1Ra.EMSA ядерных экстрактов с использованием γ- 32 P-меченой дцДНК GAS-зонда выявила быструю IFN-γ-зависимую индукцию комплекса между ядерными белками и GAS-зондом (рис. А , переулок 2). Формирование этого комплекса длилось не менее 4 часов (дорожка 3) и на него не влияли ни IL-1Ra, ни IL-1α (дорожки 4–6). В контрольном исследовании IL-1Ra отменял образование IL-1α-индуцированного комплекса ядерных белков с зондом κB (данные не показаны). Следовательно, можно сделать вывод, что пути GAS и NF-κB независимы и, следовательно, что влияние эндогенного IL-1α на противовирусный ответ IFN-γ не опосредуется сигнальным путем Jak-STAT1.
Рис. 7.Влияние IL-1 на индукцию и активацию IRF-1 IFN-γ. ( А ) ЭМС комплексов ГАЗ – СТАТ. Клетки WISH обрабатывали в течение указанного времени указанными комбинациями IFN-γ (100 единиц / мл), IL-1Ra (10 мкг / мл) и IL-1α (2 нг / мл). Были приготовлены ядерные экстракты, которые подвергались реакции с газовым зондом.( B ) Иммуноблоты IRF-1. Клетки WISH обрабатывали 2 нг / мл IL-1β в течение 1 ч, а затем 100 ед. / Мл IFN-γ. Общий клеточный экстракт получали в указанные моменты времени после добавления IFN-γ и подвергали SDS / PAGE и иммуноблоттингу с использованием антител против IRF-1 и β-актина. ( C ) EMSA комплексов IRF-1 – ISRE. Клетки WISH обрабатывали 2 нг / мл IL-1β в течение 1 часа, а затем 100 ед / мл IFN-γ в течение 30 минут. Общий клеточный экстракт подвергали EMSA с зондом ISRE. Идентичность комплекса подтверждали суперсифтингом с указанными антителами.Стрелка указывает на комплекс ISRE, а стрелка указывает на сверхсмещенные комплексы.
IRF-1 представляет собой фактор транскрипции, индуцированный IFN-γ, который опосредует многие поздние транскрипционные активности, индуцированные IFN-γ. Ранее мы продемонстрировали, что индукция IL-18BP IFN-γ отменяется у мышей с дефицитом IRF-1 (28). Мы оценили влияние экзогенного IL-1β на индукцию IRF-1 IFN-γ в клетках WISH и HaCaT. Обработка одним IFN-γ индуцировала IRF-1 только через 1 час, что было обнаружено иммуноблоттингом общего клеточного экстракта.Предварительная инкубация клеток с IL-1β в течение 1 часа перед добавлением IFN-γ не только ускоряла индукцию IRF-1, что приводило к обнаружению IRF-1 только через 30 минут, но также увеличивало его уровень (рис. В ). Активность этого повышенного IRF-1 была подтверждена наблюдением повышенного образования ядерных комплексов IRF-1 после обработки 100 ед. / Мл IFN-γ в течение 30 минут в культурах клеток с добавлением IL-1, как определено EMSA с IFN-стимулированный элемент ответа (ISRE) зонд (рис.7 C , сравните дорожки 2 и 3). Поскольку индукция мРНК IRF-1 с помощью IFN-γ ингибируется IL-1Ra (рис. A ) и усилен экзогенным IL-1, мы заключаем, что он требует активации как GAS, так и κB элементов. Этот вывод согласуется с сообщениями о присутствии элементов κB в промоторе IRF-1 (34).
Обсуждение
В настоящем исследовании мы демонстрируем, что конститутивно экспрессируемый IL-1α имеет решающее значение для индукции многих генов IFN-γ, включая HLA-DR , ICAM-1 , IL-18BP и гены, опосредующие противовирусная активность IFN-γ.В результате специфические ингибиторы IL-1α снижают противовирусную активность IFN-γ на 90% в клетках WISH человека и в аналогичной степени в эмбриональных фибробластах, полученных от двух мышей с нокаутом IL-1, по сравнению с фибробластами мышей дикого типа. . Мы также показываем, что ингибирование IL-1α и дефицит IL-1 отменяют индукцию IL-18BP in vitro и in vivo , соответственно. Примечательно, что зависимость антивирусного состояния от базального IL-1α была уникальной для IFN-γ и не наблюдалась для IFN типа I (α и β).
Мы обнаружили, что активация NF-κB базальным IL-1α необходима для индукции этих генов и для установления антивирусного состояния с помощью IFN-γ. Мы также обнаружили, что IFN-γ увеличивает экспрессию ассоциированного с мембраной IL-1α, тем самым дополнительно усиливая активацию гена парой IFN-γ / IL-1α. Наблюдение за тем, что биологическая активность или индукция гена может зависеть от нескольких сигналов, хорошо задокументировано. Такая двойная и даже множественная параллельная передача сигналов, вероятно, обеспечивает меру безопасности, типичную для многих иммунных ответов, таких как передача сигналов рецептора Т-клеток (35).
Многие исследования продемонстрировали, что TNF действует синергично с IFN-γ, вероятно, за счет активации комплементарного пути NF-κB. Здесь мы показываем, что активация NF-κB не только усиливает активность IFN-γ, но на самом деле важна для его базовой активности. Ключ к разгадке этой зависимости был предоставлен в двух более ранних исследованиях. Индукция CD40 в макрофагах и микроглиальных клетках IFN-γ ослаблялась при ингибировании конститутивно экспрессируемого TNF (36). Роль конститутивно экспрессируемого TNF в клетках микроглии, вероятно, аналогична роли IL-1α в клетках, которые мы исследовали, потому что TNF и IL-1 обладают общей способностью активировать NF-κB.В соответствии с настоящим исследованием вполне вероятно, что конститутивно экспрессируемый IL-1α поддерживал базальный уровень NF-κB, который, как сообщается, необходим для индукции CXCL9 и CXCL10 IFN-γ (17).
Критическая роль базального IL-1 в действии IFN-γ в сочетании со способностью IFN-γ увеличивать продукцию IL-1α, повышает вероятность того, что клетки, такие как кератиноциты, которые экспрессируют высокие уровни IL-1α, может повышать чувствительность инфильтрирующих макрофагов или дендритных клеток к IFN-γ, происходящему из Т-клеток.Такой каскад событий может лежать в основе хронического воспаления кожи. Роль IL-1 при псориазе была недавно подтверждена обнаружением высоких уровней IL-1-индуцируемого хемокина IL-8 в псориатических бляшках по сравнению с кожей без повреждений (37).
В заключение, это исследование показывает, что клеточный ответ на заданный сигнал определяется не только его набором рецепторов и факторов транскрипции, но также набором конститутивно экспрессируемых цитокинов и связанных с ними сигнальных молекул.
Материалы и методы
Клетки, мыши и реагенты.
Человеческие амниотические клетки WISH (CCL 25) и человеческие моноцитарные клетки THP-1 (TIB-202) были получены из Американской коллекции типовых культур (Манассас, Вирджиния). Кератиноциты человека HaCaT были получены от P. Boukamp (Немецкий центр исследования рака, Гейдельберг, Германия) (38). Гомозиготные мыши C57BL / 6 с двойным дефицитом IL-1α / β (39) были любезно предоставлены Y. Iwakura (Токийский университет, Токио, Япония).Гуманный уход за животными контролировался Комитетом по уходу и использованию животных Института Вейцмана в соответствии с руководящими принципами, установленными израильскими законами. Кроличьи поликлональные антитела к IL-1α, IL-1β, IRF-1, NF-κB, p65 и HLA-DR были от Santa Cruz Biotechnology (Санта-Крус, Калифорния). Рекомбинантные цитокины человека были от Cytolab (Реховот, Израиль). Химикаты и питательные среды были от Sigma (Реховот, Израиль).
ELISA на IL-18BP и IL-1α.
Человеческий IL-18BP измеряли в супернатантах культур методом ELISA с двумя антителами, как описано (29).Мышиный IL-18BP определяли с помощью ELISA на основе кроличьих поликлональных антител (Cytolab). IL-1α измеряли в экстрактах целых клеток с помощью ELISA с двойным антителом, приобретенного в R&D Systems (Миннеаполис, Миннесота).
Выделение РНК и ОТ-ПЦР.
клеток WISH (10 6 ) в минимальной необходимой среде (2 мл) высевали в шестилуночные планшеты на 17 часов. Различные цитокины добавляли в указанное время; затем клетки собирали и общую РНК экстрагировали с использованием реагента TRI (Sigma).кДНК получали из РНК с использованием случайных гексамеров и SuperscriptII (Invitrogen, Leek, Нидерланды). ПЦР выполняли со следующими праймерами: IL-18BP , 5′-CACGTCGTCACTCTCCTGG и 5′-CGACGTGACGCTGGACAAC; ICAM-1, , 5′-CAATGCCCAGACATCTGTGTC и 5′-CAGTGCGGCACGAGAAATTG; CXCL11 , 5′-GCTGTGATATTGTGTGCTAC и 5′-TCGATTTGGGATTTAGGCATC; IL-32 ( NK4 ), 5′-TAATGCTCCTCCCTACTTC и 5′-CAGCAGAAACTCTGGAACTG; FAT10, , 5′-GATGGCTCCCAATGCTTCCTGC и 5′-GTCACCCTCCAATACAATAAGATGC; CIITA , 5′-CTGAAGGATGTGGAAGACCTGGGAAAGC и 5′-GTCCCCGATCTTGTTCTCACTC; HLA-DR , 5′-GAGTTTGATGCTCCAAGCCCTCTCCCA и 5′-CAGAGGCCCCCTGCGTTCTGCTGCATT; Mx1 , 5′-CATCGACCTCATTGACTCCC и 5′-TCCACATTACTGGGGACCAC; Mx2 , 5′-TCCAAAAATCGCTCCCGTTG и 5′-GACGTTTGCTGGTTTCCAAG; ISG20, , 5′-GTTGCAGCCTCGTGAACGTC и 5′-CTGTGTCCAAGCAGGCTGTTC; β- Актин , 5′-GTGGGGCGCCCCAGGCACCA и 5′-CTCCTTAATGTCACGCACGATTTC.
Амплификации проводили путем начальной денатурации при 92 ° C в течение 2 минут и 28 циклов денатурации при 92 ° C в течение 30 секунд, отжига при 62 ° C в течение 45 секунд, удлинения при 72 ° C в течение 45 секунд и окончательного удлинения при 72 ° C. ° C в течение 10 мин. Полученные продукты ПЦР разделяли электрофорезом в агарозном (1%) геле.
Анализ массива ДНК.
Клетки WISH человека обрабатывали 100 ед. / Мл IFN-γ, 10 мкг / мл IL-1Ra или их комбинацией в течение 3, 6 и 17 часов.РНК выделяли и подвергали анализу массива экспрессии с использованием генных чипов Affymetrix U133A. Полученные файлы данных Excel были отсортированы по следующему алгоритму: были отобраны гены, которые присутствовали и индуцировались IFN-γ в ≥4 раза. Из этих генов окончательный отбор делали те, чей сигнал после комбинированной обработки IFN-γ и IL-1Rα был снижен на ≤2 / 3 по сравнению с сигналом от клеток, обработанных одним IFN-γ. Гены, базальный уровень которых был снижен одним только IL-1Ra на ≤2 / 3, также были исключены.
Противовирусный анализ.
клеток WISH (40000 / мл) высевали в 100 мкл MEM в 96-луночные планшеты. Клетки инкубировали с серийно разведенными в 2 раза запасами IFN-α2, IFN-β или IFN-γ в течение 17 часов. Вместе с IFN добавляли антитела к IL-1β, TNF-α, IL-1Ra, анти-IL-1α или анти-IL-β. Вирус везикулярного стоматита добавляли через 17 часов, а защиту от вирусного цитопатического эффекта оценивали в лунках, окрашенных кристаллическим фиолетовым, через 40 часов.
Люциферазных репортерных векторов.
Люциферазный репортерный вектор pGL3 (656), содержащий плазмидный вектор pGL3-Basic (Promega, Madison, WI), в который лигирован участок промотора человеческого IL-18BP длиной 656 пар оснований, был описан ранее (28). Элемент κB-response мутировали с получением вектора pmGL3 (656) с помощью метода мутагенеза на основе ПЦР (40), который использовал следующие смысловые праймеры и его комплементарные праймеры: 5′-CAACTTGG AAAATTTT AGTGCCTAGGAAAG. Мутировавшие области выделены курсивом.
Анализ временной трансфекции.
клеток HaCaT или WISH в шестилуночных планшетах (5 × 10 5 клеток на лунку) трансфицировали FuGene 6 (Roche Molecular Biochemicals, Mannheim, Германия), указанным репортерным вектором люциферазы (0,5 мкг на лунку) и pSV40. βGAL (0,2 мкг на лунку; Promega). Через 16 часов клетки обрабатывали 100 ед. / Мл IFN-γ, 10 мкг / мл IL-Ra или их комбинацией в бессывороточной среде в течение 24 часов. Затем клетки собирали и лизировали и измеряли активность люциферазы. Все результаты были нормализованы относительно активности β-галактозидазы.
EMSA.
Следующие олигонуклеотиды были использованы для создания зондов: 5′-GATCCGTCGACATTTCCCGTAAATCG и 5′-AGCTTGATTTACGGGAAATGTCGACG, соответствующие GAS, и 5′-AGTTGAGGGGACTTTCCCAGGC и 5′-CCTCCGGACT, соответствующие сайту связывания GAAAGTCCG, с сайтом связывания GAAAGTCCCCG с сайтом связывания GAAAGTCCCCGCGC и 5′-CCTCCG. дцДНК (10 пмоль) метили [γ- 32 P] АТФ и использовали в EMSA, как описано ранее (28).
Статистика.
Достоверность различий между группами, показывающими схожую дисперсию, оценивали с помощью теста Стьюдента t .
Благодарности
Мы благодарим Сару Барак за отличную техническую помощь, Ширли Хорн Сабан за выполнение анализа массива генов Affymetrix и Йохиро Ивакура за предоставление мышей с двойным нокаутом по IL-1α / β. Это исследование было поддержано Serono Group.
Сноски
- ‡ Кому следует направлять корреспонденцию. Электронная почта: menachem.rubinstein {at} weizmann.ac.il
Вклад авторов: V.H., C.A.D. и M.R., разработанные исследования; V.H., D.N. и A.W. проведенное исследование; Д.Н. предоставил новые реагенты / аналитические инструменты; V.H., C.A.D., D.N. и M.R. проанализировали данные; и М.Р. написали статью.
Авторы заявляют об отсутствии конфликта интересов.
- Сокращения:
- IRF,
- Регуляторный фактор IFN;
- GAS,
- γ-активированная последовательность;
- MEF,
- эмбриональный фибробласт мыши;
- IL-1Ra,
- антагонист рецептора IL-1;
- IL-18BP,
- IL-18-связывающий белок;
- PDTC,
- пирролидиндиокарбамат;
- ISRE,
- IFN-стимулированный ответный элемент.
Свободно доступен онлайн через опцию открытого доступа PNAS.
- © 2007 Национальная академия наук США
Различные роли IFN в сенсибилизации к эндотоксиновому шоку вирусом, повышающим лактатдегидрогеназу | Международная иммунология
Аннотация
Было проанализировано влияние заражения мышей вирусом, повышающим уровень лактатдегидрогеназы (LDV), обычно непатогенным вирусом, на сопутствующий бактериальный эндотоксиновый шок с точки зрения летальности и продукции цитокинов.Сильное повышение восприимчивости к шоку наблюдалось у мышей, остро инфицированных этим вирусом. Это коррелировало с резким увеличением выработки фактора некроза опухоли и фактора ингибирования лейкемии и контролировалось генетическим фоном мышей. Вирусная инфекция привела к дисбалансу цитокинового ответа на LPS с усилением провоспалительных цитокинов, включая IL-18 и IFN-γ, и замедленной секрецией противовоспалительного IL-10, что могло привести к усилению активации макрофагов.Повышенная продукция IFN-γ была связана с вирус-индуцированной восприимчивостью к шоку. В отличие от других вирусных инфекций, IFN-α / β уменьшал продукцию IFN-γ и, как следствие, увеличивал ответ на LPS у LDV-инфицированных животных.
Введение
Было показано, что как генетические факторы, так и факторы окружающей среды определяют исход сепсиса и септического шока. Среди этих факторов коинфекция вирусами, такими как грипп или респираторно-синцитиальный вирус, в значительной степени увеличивает как частоту, так и тяжесть бактериальных инфекций (1).Экспериментальные модели показали повышенную восприимчивость мышей к сопутствующему воздействию ЛПС и инфекции, вызываемой возбудителями, включая вирус лимфоцитарного хориоменингита (LCMV), аденовирус, вирус Тейлера или вирус везикулярного стоматита (2–6). Сообщается, что ИФН типа I и, в меньшей степени, типа II являются основными медиаторами обострения эндотоксинового шока вирусами (2–5). Однако, поскольку эти вирусы являются патогенными сами по себе или могут, по крайней мере, вызывать патологию при соответствующих обстоятельствах, трудно отличить обострение индуцированной ЛПС патогенности вирусными агентами и между усилением или индукцией вирусной патологии. через механизмы, запускаемые эндотоксином.
Вирус, повышающий уровень лактатдегидрогеназы (LDV), представляет собой артеривирус, непатогенный per se для большинства линий инфицированных мышей, поскольку у этих животных его тропизм ограничен субпопуляцией нежизнеспособных макрофагов. Однако LDV вызывает быстрый, но временный всплеск продукции цитокинов, который может мешать текущим патологическим механизмам, изначально не связанным с инфекцией [см. Обзор (7)]. Таким образом, он представляет собой уникальную модель для анализа последствий сопутствующей вирусной инфекции для цитокин-опосредованных заболеваний, таких как эндотоксиновый шок.Здесь мы сообщаем, что LDV, как и другие вирусы, резко увеличивает восприимчивость инфицированных хозяев к LPS-шоку. Эта сенсибилизация, контролируемая генетическими факторами, коррелирует с несбалансированной выработкой провоспалительных цитокинов и противовоспалительного IL-10. Это вызванное вирусами обострение эндотоксинового шока частично опосредуется IFN-γ, но сильно усиливается у животных, не реагирующих на IFN типа I.
Методы
Животные
129 / Sv, BALB / C и DBA / 2 были выведены в Институте исследования рака Людвига (Брюссель) Дж.Warnier и используется в возрасте 8–12 недель. Мышей с дефицитом рецептора IFN-γ (G129), любезно предоставленных Ф. Бромбахером (Институт иммунобиологии Макса Планка, Фрайбург, Германия), выращивали таким же образом, как и животных 129 / Зв, от которых они были первоначально получены с помощью S. Хуанг и М. Аге (8). Мыши IFN-α / βR 0/0 также были первоначально получены M. Aguet (9). Проект одобрен местной комиссией по уходу за животными.
Вирус
Мышей инфицировали внутрибрюшинно (т.е.p.) инъекции ~ 2 × 10 7 50% инфекционных доз (ID 50 ) LDV (штамм Riley; American Type Culture Collection, Rockville, MD, USA) в 500 мкл физиологического раствора.
ЛПС
LPS из Escherichia coli ( E. coli 0111: B4; Sigma, Bornem, Бельгия) вводили внутрибрюшинно. в 500 мкл физиологического раствора. Чувствительность к эндотоксиновому шоку определяли путем инъекции группам мышей титрованных доз ЛПС. LD 50 рассчитывали по методу Рида и Мюнча (10).
Анализ цитокинов
Фактор некроза опухоли (TNF) определяли с помощью биоанализа, как описано ранее (11, 12). Вкратце, клетки клона 13 WEHI-164 инкубировали в присутствии актиномицина D с образцами для тестирования в среде, дополненной LiCl, чтобы максимизировать чувствительность к TNF-опосредованной цитотоксичности. Затем жизнеспособность клеток оценивали с помощью колориметрического анализа на основе тетразолия и рассчитывали концентрации TNF в отношении максимальной летальности клеток, полученной с помощью рекомбинантного цитокина (любезный подарок W.Фирса, Гентский университет, Бельгия). 1 Ед. Мл -1 была произвольно определена как концентрация TNF, которая индуцировала 50% -ную летальность клеток (что соответствует ± 1 мкг / мл -1 ).
IFN-γ измеряли с помощью ELISA с использованием коммерческого набора (CytoSets®), следуя рекомендациям производителя (Biosource, Camarillo, CA, USA). IL-12 и фактор ингибирования лейкемии (LIF) анализировали с помощью наборов Quantikine® (R&D Systems, Миннеаполис, Миннесота, США). IL-1β и IL-10 измеряли с использованием улавливающих и детектирующих антител и стандартов из наборов DuoSet® (R&D Systems) со стрептавидином-HRP от Amersham (Литл-Чалфонт, Великобритания) и Ultra-TMB-ELISA® от Pierce (Рокфорд, Иллинойс, США). СОЕДИНЕННЫЕ ШТАТЫ АМЕРИКИ).Набор ELISA от MBL (Нагоя, Япония) использовали для измерения уровней IL-18.
Концентрации IL-22 в плазме измеряли с помощью ELISA с использованием мышиных mAb, направленных против человеческого IL-22 (Mh32B2), и аффинно очищенных поликлональных кроличьих антител против мышиного IL-22. Mh32B2 перекрестно реагирует с мышиным IL-22. Вкратце, микротитрационные планшеты (Immunoplates, Nunc, Roskilde, Дания) покрывали Mh32B2 (5 мкг мл -1 ) в 20 мМ глицине, 30 мМ NaCl (pH 9,2) буфере и инкубировали в течение ночи при 4 ° C. Затем планшеты промывали PBS с Tween 20 (5 × 10 -4 ) и блокировали PBS-BSA (5%) в течение 1 ч при 37 ° C.После промывки в планшеты добавляли разведения образцов в PBS с 2,5% BSA, которые инкубировали в течение 3 ч при 37 ° C. Биотинилированное кроличье поликлональное антитело против mIL-22 (0,136 мкг / мл -1 ) использовали в качестве вторичного антитела в течение 2 ч инкубации при 37 ° C. Затем планшеты промывали и замачивали в течение 7 минут в NaCl, содержащем 1% NP-40 (Fluka AG, Buchs, Швейцария). Связывание второго антитела детектировали с помощью стрептавидин-HRP (Amersham). Анализ был разработан путем добавления 50 мкл Ultra-TMB-ELISA® (Pierce), и реакцию останавливали с помощью 50 мкл 2 M H 2 SO 4 перед измерением оптической плотности при 450 нм.
IL-6 анализировали путем инкубации серийных разведений образцов с IL-6-зависимой B-клеточной гибридомой 7TD1 (2000 клеток на микролунку) в 0,2 мл среды Искова, содержащей 10% FCS и дополненной 0,24 мМ L-аспарагина, 0,55 мМ L-аргинин, 1,5 мМ L-глутамин, 0,05 мМ 2-меркаптоэтанол, 0,1 мМ гипоксантин и 0,016 мМ тимидин. Жизнеспособность оценивали через 4 дня с помощью определения гексозаминидазы. Результаты, выраженные в Ед / мл -1 , были определены как концентрация, обеспечивающая полумаксимальный рост клеток (13).
Антитело
Козья антисыворотка против TNF была приготовлена в Гормонологической лаборатории Centre d’Economie Rurale (Марлой, Бельгия) после первой иммунизации 20 мкг рекомбинантного мышиного TNF с последующими ежемесячными иммунизациями 10 мкг TNF. Поликлональные антитела получали осаждением сульфатом аммония.
Проточная цитометрия
Популяции макрофагов анализировали проточной цитометрией, как описано (14), с флуоресцеинированным mAb против F4 / 80 мыши (Serotec, Raleigh, NC, USA).
Статистический анализ
При необходимости статистический анализ проводился с использованием непараметрического непарного двустороннего критерия Манна – Уитни.
Результаты
Обострение патогенности эндотоксинов в результате инфекции LDV
Выживаемость либо неинфицированных мышей 129 / Sv, либо животных, остро инфицированных LDV, контролировали после введения титрованных доз LPS. Как показано на рис. 1, неинфицированные мыши были довольно устойчивы к эндотоксиновому шоку.Напротив, животные, инфицированные LDV, демонстрировали резко повышенную чувствительность к LPS. Эта повышенная восприимчивость к эндотоксиновому шоку наблюдалась у мышей, инфицированных LDV в течение 1 и 2 дней, но не в такой степени у животных, инфицированных в течение 4 или 7 дней (данные не показаны).
Рис. 1.
Повышение восприимчивости мышей к эндотоксиновому шоку при заражении LDV. Выживаемость контролировали после инъекции титрованных доз LPS группам из шести мышей 129 / Sv, через 24 часа после введения физиологического раствора или LDV.
Рис. 1.
Повышение восприимчивости мышей к эндотоксиновому шоку при заражении LDV. Выживаемость контролировали после инъекции титрованных доз LPS группам из шести мышей 129 / Sv, через 24 часа после введения физиологического раствора или LDV.
Поскольку TNF играет основную роль в развитии эндотоксинового шока, было проанализировано влияние инфекции LDV на его продукцию в ответ на введение LPS. Наши результаты показали синергетический эффект ЛПС и ЛДВ на секрецию этого цитокина (рис.2А). Инфекция LDV привела к очень низким титрам TNF с уровнями в плазме ниже 100 Ед. Мл -1 у всех протестированных мышей через 1 день после заражения. Через 2 часа после введения 24 мкг LPS у неинфицированных мышей развился умеренный ответ TNF. Напротив, уровни TNF были резко увеличены у LDV-инфицированных животных, которые получали такую же дозу LPS. Затем уровни TNF быстро снизились и достигли гораздо более низких значений через 6 часов после введения LPS (таблица 1).
Таблица 1.Кинетика продукции цитокинов у мышей, инфицированных LDV, в ответ на LPS
| Cytokinea | LDV | Время после введения LPS (часы) b | P valuec33 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 933 933 933 933 | 2 | 6 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TNF | — | 884 ± 143 | 20 ± 2 | 0.0079 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| + | 97 576 ± 38 517 | 2483 ± 954 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| IL-1β | — | 91 ± 30 | 66 ± 22 | 0,0079 | ± | 933 17 | 444 ± 103 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LIF | — | 127 ± 69 | 129 ± 54 | 0,0079 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| + | 2373 ± 335 | 14 347 ± 1054 933 9333 9333 | 14 347 ± 1054 933 36 | — | 12 602 ± 5492 | 2118 ± 845 | 0.0317 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| + | 104721 ± 18 054 | 327523 ± 102 884 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| IFN-γ | — | NDd | 230 ± 80 | 0,3657 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7002 ± 1099 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ИЛ-12 | — | 332 ± 98 | 484 ± 113 | 0,0556 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| + | 852 ± 262 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 852 ± 262 |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||